蜂蜜中氯霉素殘留量的不確定度評定
2020-08-12 12:17:10楊濤,袁輝
廣州化工 2020年14期
關鍵詞:標準
楊 濤,袁 輝
(新疆產品質量監督檢驗研究院,新疆 烏魯木齊 830011)
蜂蜜因含有豐富的果糖、葡萄糖、多種維生素、有機酸等對人體有益的礦物質,被譽為大自然中最完美的營養食品。氯霉素是一種強力抗生素,能有效控制蜂群感染,減少蜜蜂死亡情況,但如果過量使用氯霉素,會導致部分氯霉素在蜂蜜中殘留。氯霉素作為一種廣譜高效抗生素,有嚴重的副作用,它人類的毒性較大,因此,中華人民共和國農業部公告第235號《動物性食品中獸藥最高殘留限量》中嚴格規定動物性食品中氯霉素不得檢出。目前,蜂蜜中氯霉素的檢測標準主要有:微生物法、酶聯免疫法、氣相色譜-質譜法、液相色譜-串聯質譜法。目前報道的有關蜂蜜中氯霉素殘留量不確定度評定主要依據SN/T 1864-2007 《進出口動物源食品中氯霉素殘留量的檢測方法 液相色譜-串聯質譜法》[1],而國內大多數檢測機構采用的檢測方法是GB/T 18932.19-2003 《蜂蜜中氯霉素殘留量的測定方法 液相色譜-串聯質譜法》[2],本文作者結合前期不確定度研究成果[3],現根據此檢驗方法分析蜂蜜中氯霉素殘留量不確定度的來源。
1 實 驗
1.1 原 理
用乙酸乙酯提取其中的待測組分,濃縮后用水溶解并過HLB小柱凈化,液相色譜-串聯質譜儀測定,外標法定量。
1.2 主要儀器與試劑
TSQ GUANTIS高效液相色譜-質譜/質譜儀,美國thermofisher公司;電子天平(感量0.1 mg,感量0.01 g),德國Sartorius公司;固相萃取裝置,美國J2 Scientific公司;離心機,上海安亭。
氯霉素標準品(99%),德國 Dr公司;乙腈、甲醇、乙酸乙酯(色譜純),德國默克公司;其它試劑為分析純;超純水由美國millipore純水儀制得;蜂蜜樣品為市場中購買。
1.3 檢測方法
1.3.1 提 取
準確稱取5 g 試樣(精確到0.01 g)于50 mL 離心管中,加入5 mL 水,搖勻使其溶解;加15 mL乙酸乙酯提取,離心后準確吸收取12 mL乙酸乙酯溶液進行濃縮,盡干,用5 mL 水溶解待凈化。
1.3.2 凈 化
用5 mL乙酸乙酯洗脫。洗脫液在50 ℃下用氮吹儀濃縮至近干,用1 mL乙腈水溶液(2+8)溶解殘渣,用0.22 μm濾膜過濾后上儀器測定。
1.3.3 色譜條件
流動相為乙腈:水溶液(2∶8),流速0.3 mL/min,柱溫40 ℃,進樣量:5 μL。質譜條件:電噴霧負離子模式,多反應監測模式(MRM)。
1.4 不確定度來源及數學模型
根據檢驗方法,不確定度主要來源如下:一是標準溶液引入的不確定度,包含標準品純度、稱量、標準溶液配制以及標準曲線擬合等分量;二是樣品前處理過程引入的不確定度,包括樣品的稱量、定容以及待測組分提取回收率等分量。
依據標準檢驗方法和評估指南[4]本試驗中氯霉素計算數學模型為:
式中:X——試樣中氯霉素的含量,μg/kg
m——試樣質量,g
V——試樣提取液總體積,mL
c——從標準曲線上測得樣品中氯霉素的質量濃度,ng/mL
f——樣品中待測組分回收率,%
2 不確定度評定
2.1 標準溶液引入的不確定度
2.1.1 標準品純度引入的不確定度
根據標準品證書提供的內容,氯霉素純度大于99%,按照矩形分布,其相對不確定度為:
2.1.2 標準品稱量引入的不確定度
當稱取氯霉素標準品10 mg時所使用的天平最大允許誤差為±0.1 mg,按照矩形分布,其相對不確定度為:
urel(s) =0.058/10=0.0058
2.1.3 標準溶液稀釋配制過程中由移液器和玻璃量器引入的不確定度
系列標準溶液的稀釋配制要使用不同規格的移液器和玻璃量器,依據JJG 646-2006《移液器檢定規程》[5]、JJG 196-2006《常用玻璃量器檢定規程》[6],其引入的不確定度見表1。

表1 校準玻璃量器和移液器引入的不確定度
根據表1中的數據,結合各自使用的次數,則移液器和玻璃量器引入的相對不確定度為:
2.1.4 標準溶液稀釋過程中溫度引入的不確定度
在標準溶液稀釋配制過程中,環境溫度控制在(20±2)℃,標準溶液是用乙腈稀釋配制,其在20 ℃時的膨脹系數為1.37×10-3mL/℃,依據矩形分布,移液器和玻璃量器由于溫度校準引入的不確定度見表2。
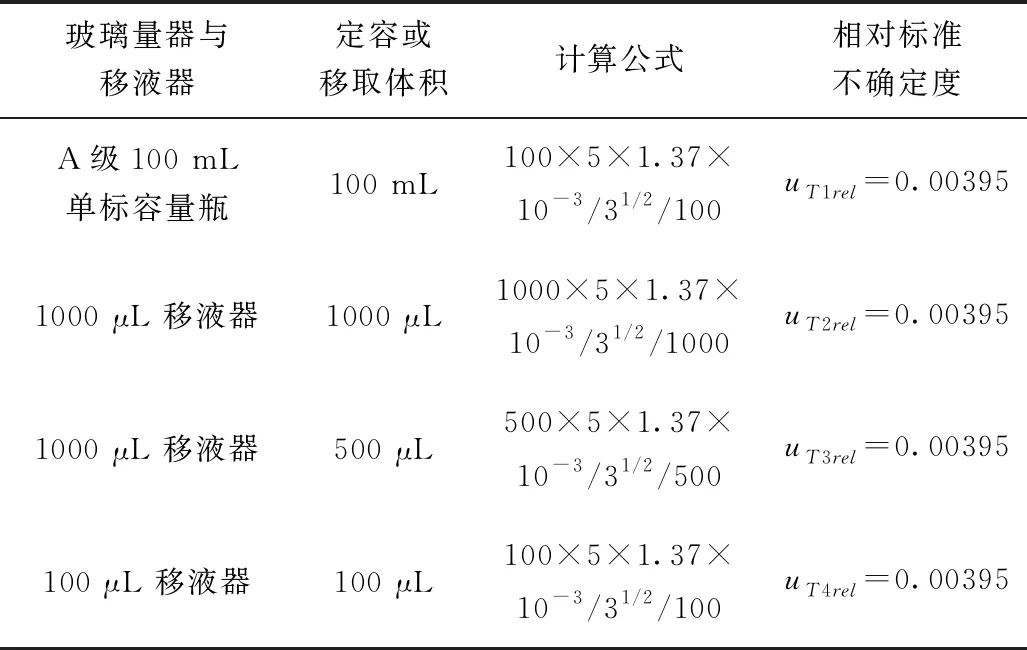
表2 溫度校準引入的不確定度
由表2中的數據結合使用次數,由于溫度變化引入的相對不確定度:
2.1.5 最小二乘法擬合標準曲線的不確定度
將標準溶液稀釋為1、2、5、10、20 ng/mL五個梯度濃度,每個濃度上機測定3次,即n=15,采用最小二乘法對標準溶液濃度/平均峰面積對應的曲線擬合,得到其相對不確定度。標準溶液系列質量濃度與對應峰面積的數據見表3。
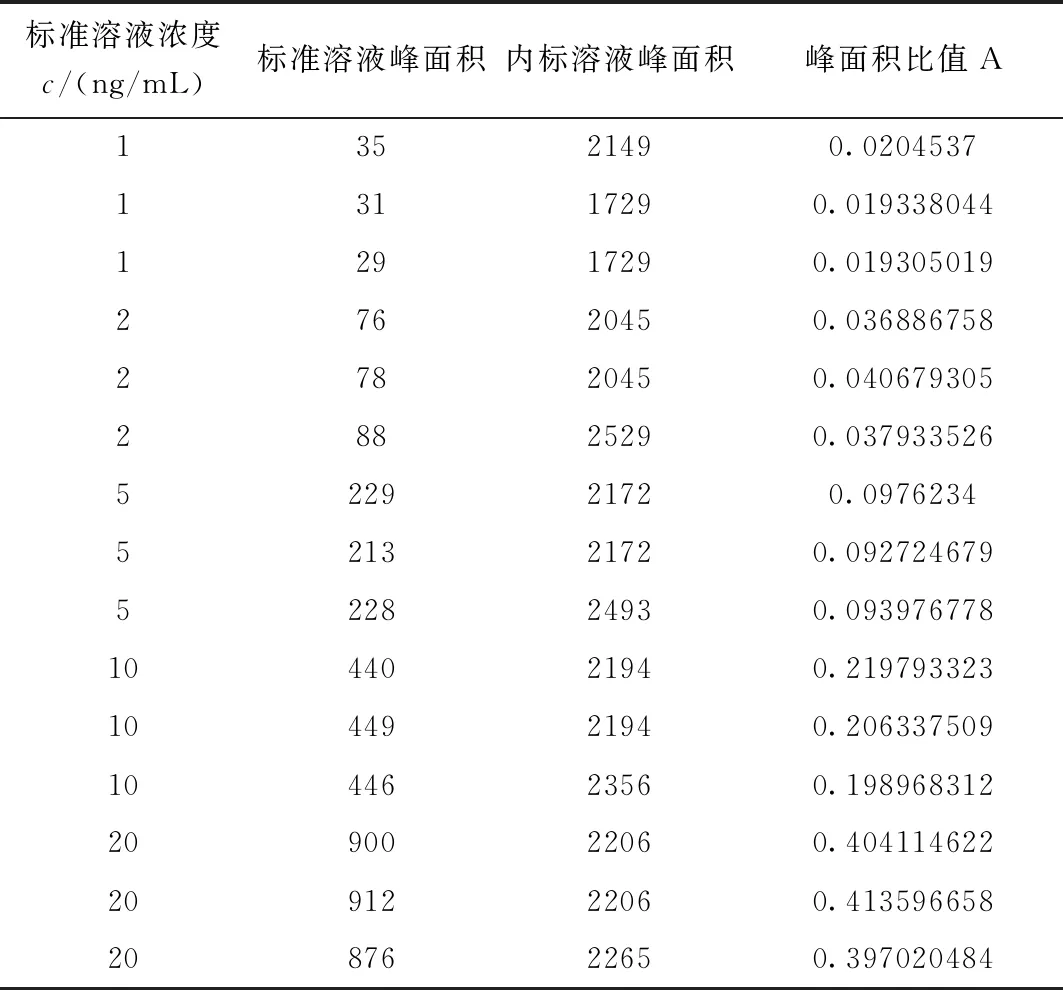
表3 液相色譜-串聯質譜法測定氯霉素的標準曲線數據
對樣品處理液中的氯霉素共測量6次,即p=6。測得樣品處理液中氯霉素的平均含量為x=9.3163 ng/mL。于是由最小二乘法擬合標準曲線所引入的不確定度u(q)為:
其中:

標準曲線擬合引入的相對不確定度為:
urel(q)=u(q)/x=0.0402
因此,在標準溶液配制測定過程中,各分量合成的不確定度為:
2.2 樣品前處理過程引入的不確定度
2.2.1 樣品稱量過程引入的不確定度
當稱取蜂蜜5.00 g時,其所使用的天平允許最大誤差為±0.01 g,根據矩形分布,其相對不確定度為:
urel(m)=0.00577/5.00=0.00115
2.2.2 樣品前處理過程體積引入的不確定度
樣品前處理過程中使用15 mL單標線吸量管1次,10 mL單標線吸量管1次,1 mL單標線吸量管1次,樣品前處理過程引入的相對不確定度見表4。
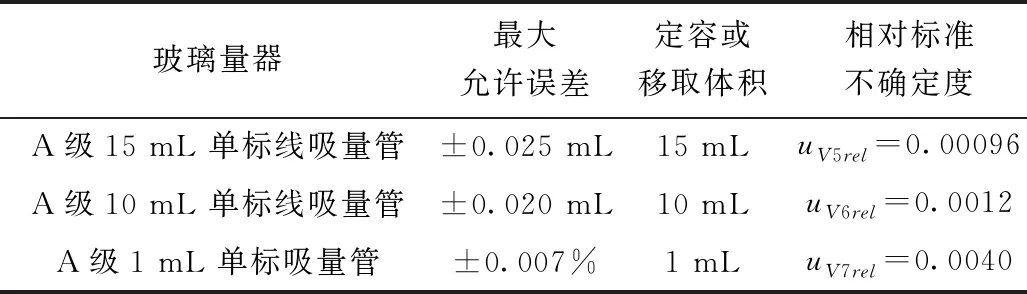
表4 樣品前處理過程中玻璃量器引入的不確定度
由表4中的數據,合成前處理過程中玻璃量器引入的相對不確定度為:
2.2.3 樣品前處理回收率引入的不確定度
對陰性樣品添加10 ng/mL的氯霉素,進行6次加標回收實驗,回收率分別為92.9%,93.3%,92.9%,92.7%,93.8%,93.1%,平均回收率為93.16%,相對標準偏差為0.392%,則由回收率引入的相對不確定度為:
urel(f)=0.00392
2.3 不確定度合成
2.3.1 標準不確定度合成
由以上分析過程各相對不確定度分量見表5。

表5 各相對不確定度分量匯總表
由上述各不確定度分量的數據合成標準不確定度為:
2.3.2 擴展不確定度
依據JJF 1135-2005《化學分析測量不確定度評定》[7],在95%的置信水平下,取k=2,蜂蜜樣品中氯霉素的測量值為9.3163 ng/mL,結合前處理過程,計算得到樣品中氯霉素的含量為2.79 μg/kg,則擴展不確定度為:
U=2.79×0.0502×2=0.280 μg/kg
2.3.3 不確定結果
按照該方法測定蜂蜜中氯霉素的結果為:X=(2.79±0.28) μg/kg,k=2。
3 結 論
根據實驗過程,對蜂蜜中氯霉素殘留量測定的不確定度來源進行了評定,評定結果表明:影響的主要因素是采用最小二乘法標準曲線擬合對氯霉素質量濃度的影響,而樣品稱量、前處理過程中等引入的不確定度基本可忽略不計。因此建議在采用該方法的檢測過程中,要對標準溶液配制步驟以及標準曲線擬合過程進行強化控制,使檢測結果準確有效。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
當代陜西(2019年8期)2019-05-09 02:22:48
上海建材(2019年1期)2019-04-25 06:30:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
專用汽車(2016年4期)2016-03-01 04:13:43
質量與標準化(2015年9期)2015-12-31 11:41:40
中國質量與標準導報(2014年4期)2014-03-11 19:54:25
中國質量與標準導報(2014年10期)2014-02-28 22:25:47
中國質量與標準導報(2014年7期)2014-02-28 22:24:39
