阿膠口服液中粗多糖含量測定方法的研究
2020-08-05 09:59:42劉春霖吳曉云謝強勝高天陽董蓬
藥學研究 2020年7期
關鍵詞:方法
劉春霖,吳曉云,謝強勝,高天陽,董蓬
(山東省食品藥品檢驗研究院,山東省食品藥品安全檢測工程技術研究中心,山東 濟南 250101)
阿膠為馬科動物驢(EquusasinmL.)的干燥皮或鮮皮經煎煮、濃縮制成的固體膠。它是我國傳統的名貴補血中藥,具有補血滋陰,潤燥,止血,增強體質,增強免疫力等功效[1-3]。本次研究的阿膠口服液是以阿膠為主要原料,添加熟地黃、黨參、枸杞子、黃芪等中藥材,經提取、濃縮、配制、灌裝等主要工藝制成的產品,具有增強免疫力的保健功能。上述添加的中藥材均含有多糖類成分,文獻研究報道具有免疫調節、抗氧化、抗疲勞的作用[4-8]。近年來,很多學者對不同中藥材的粗多糖含量進行了研究[9-10],但是針對阿膠口服液中粗多糖的研究還比較少。本研究采用沉淀蛋白質等前處理方法,使樣品中相對分子質量大于10 000的高分子物質與阿膠分離,然后在體積分數80%乙醇溶液中沉淀,與水溶液中單糖和低聚糖分離,用苯酚-硫酸反應,其呈色強度與溶液中糖的濃度成正比,在485 nm波長下比色測定粗多糖(以葡聚糖計)含量。
1 儀器與試藥
1.1 儀器 Mettler Toledo Ms十萬分之一電子天平(德國梅特勒公司);恒溫水浴鍋(上海樹立儀器儀表有限公司);TDZ5-WS離心機(長沙湘儀離心機儀器有限公司);XK96-B渦旋混合器(姜堰市新康醫療器械有限公司);TU-1901紫外分光光度計(北京普析通用公司)。
1.2 試劑試藥 乙酸鋅、亞鐵氰化鉀、苯酚、無水乙醇、濃硫酸,分析純;水為去離子水;乙酸鋅溶液:稱取乙酸鋅[Zn(CH3COO)2·2H2O]21.9 g,加冰乙酸3 mL,加水溶解并稀釋至100 mL;亞鐵氰化鉀溶液:稱取亞鐵氰化鉀{K4[Fe(CN)6]·3H2O}10.6 g,加水溶解并稀釋至100 mL;80%(V/V)乙醇溶液:20 mL水中加入無水乙醇80 mL,混勻;苯酚溶液(50 g·L-1):稱取精制苯酚5.0 g,加水溶解并稀釋至100 mL,混勻,備用。
葡聚糖標準品:相對分子量500 000,批號:BCBN7863V,購自Sigma公司。
葡聚糖標準儲備液:準確稱取干燥至恒重的葡聚糖對照品0.5 g,加水溶解,并定容至50 mL,混勻,置冰箱中保存。此溶液1 mL含葡聚糖10 mg。
葡聚糖標準使用液:吸取葡聚糖標準儲備液1.00 mL,置100 mL容量瓶中,加水至刻度,混勻,置冰箱中保存。此溶液1 mL含葡聚糖100 μg。
阿膠:某阿膠制品公司提供,和樣品來源為同一家公司。
樣品:某阿膠制品公司提供,3批編號分別為1、2、3。
2 方法與結果
2.1 標準曲線系列溶液制備 精密吸取葡聚糖標準使用液0.00、0.10、0.20、0.40、0.60、0.80、1.00、1.20、1.40、1.60 mL(相當于葡聚糖0、10、20、40、60、80、100、120、140、160 μg),分別置于25 mL比色管中,準確加水補充至2.0 mL,加入50 g·L-1苯酚溶液1.0 mL,在旋轉混合器上混勻,小心加入濃硫酸10.0 mL,于旋轉混合器上小心混勻,置沸水浴中煮沸2 min,冷卻后用分光光度計在485 nm波長處,以試劑空白為試驗參比,1 cm比色皿測定吸光度值。以葡聚糖濃度(μg)為橫坐標,吸光度值為縱坐標,繪制標準曲線。
2.2 樣品溶液的制備
2.2.1 試樣沉淀蛋白質處理 取混合均勻的試樣15 mL,置250 mL容量瓶中,加水50 mL,緩慢加入乙酸鋅溶液5 mL和亞鐵氰化鉀溶液5 mL,加水至刻度,混勻,靜置30 min,用干燥濾紙過濾,取續濾液備用。
2.2.2 沉淀粗多糖 準確吸取上一步濾液5 mL,置于50 mL離心管中,加入無水乙醇20 mL,混勻,置4 ℃冰箱中冷藏過夜,以4 000 rpm離心30 min,棄去上清液,殘渣用體積分數80%乙醇溶液數毫升洗滌,以4 000 rpm離心15 min,離心后棄上清液,反復操作2次,殘渣用水溶解并定容至25 mL,混勻,此溶液為樣品測定液。
2.2.3 樣品測定 分別吸取2.0 mL樣品測定液置25 mL比色管中,加入50 g·L-1苯酚溶液1.0 mL,混勻,加入濃硫酸10.0 mL,混勻,密塞,沸水浴加熱2 min,立即冷卻至室溫得待測溶液。在485 nm波長處測定吸光度值。從標準曲線上查出葡聚糖的質量,計算,即得樣品粗多糖含量。
2.3 標準曲線回歸方程線性范圍 取標準曲線系列溶液進儀器測定,結果表明葡聚糖的標準溶液在10.8~172.8 μg范圍內具有良好的線性關系,相關系數為0.999 9,結果見表1。

表1 線性試驗結果
2.4 取樣量考察 分別量取同一批號的樣品5、10、15、25 mL各2份,按上述樣品溶液的制備方法制得待測溶液,測得葡聚糖含量分別為244.6、253.1、285.7、280.9 mg·(100 mL)-1,結果表明取樣量為15 mL時蛋白質沉淀完全,粗多糖沉淀量適當,樣品溶液吸光度值在標準曲線中段,所以確定15 mL為取樣量。
2.5 重復性考察 精密量取同一批號混合均勻的樣品6份,按上述方法制得待測溶液,比色測定后計算葡聚糖含量,結果6份樣品葡聚糖含量的RSD為1.4%,表明該方法重復性良好。
2.6 精密度考察 取制得的待測溶液,按上述方法連續測定6次吸光度,利用標準曲線計算葡聚糖含量,結果6次葡聚糖含量的RSD為0.11%,表明該方法精密度良好。
2.7 穩定性考察 取制得的待測溶液,按上述方法分別于比色后0、1、2、4 h測定吸光度,結果4次葡聚糖含量的RSD為0.96%,表明比色后供試品溶液在室溫條件下4 h內穩定。
2.8 回收率考察 精密量取同一批號混合均勻的樣品9份,分為3組濃度,每組濃度3份樣品,每組分別精密稱取葡聚糖對照品約18、22.5、27 mg,置250 mL容量瓶中,分別精密加入混勻的試樣7.5 mL,依法制備加標樣品。按上述方法進行測定,結果平均回收率在97.1%~100.1%,表明該方法回收率好,結果見表2。
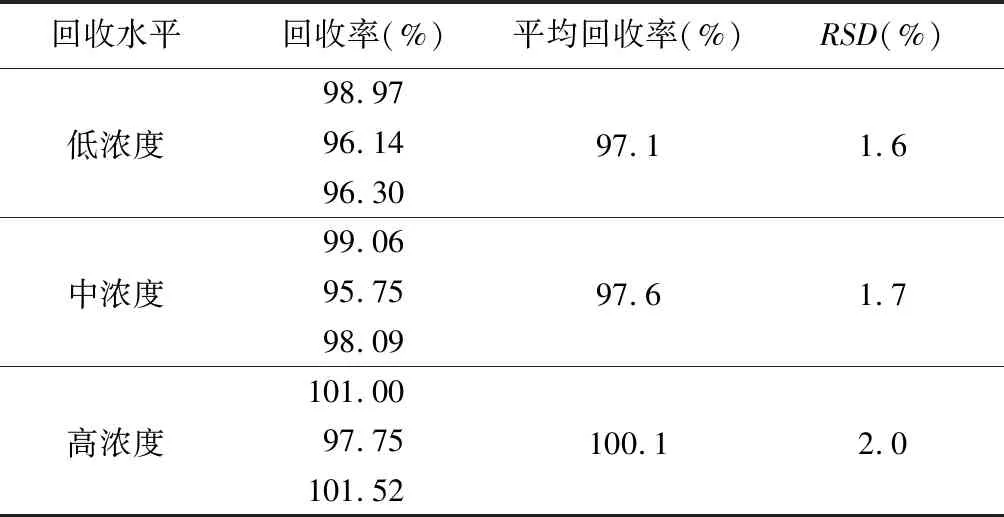
表2 回收率試驗結果(n=3)
2.9 方法檢出限 參照GB/T 5009.1-2003《食品衛生檢驗方法理化部分總則》中附錄A.2.2規定,檢出限為3倍空白值的標準偏差(測定次數n≥20)相對應的質量,吸光法按國際理論與應用化學家聯合(IUPAC)規定。檢出限按下式進行計算:檢出限=Ks/b;b-標準曲線回歸方程中的斜率;s-20次空白值的標準偏差;K-一般為3,結果見表3。
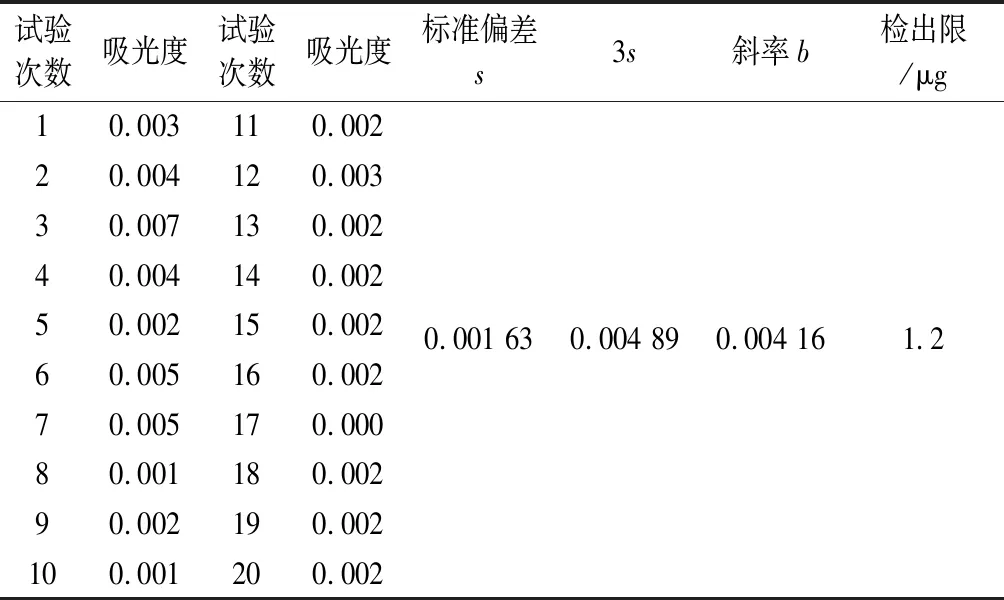
表3 檢出限試驗結果
2.10 樣品測定
2.10.1 沉淀蛋白質方法 精密量取3批混合均勻的樣品15 mL,按上述方法進行測定,計算葡聚糖含量,結果見表4。
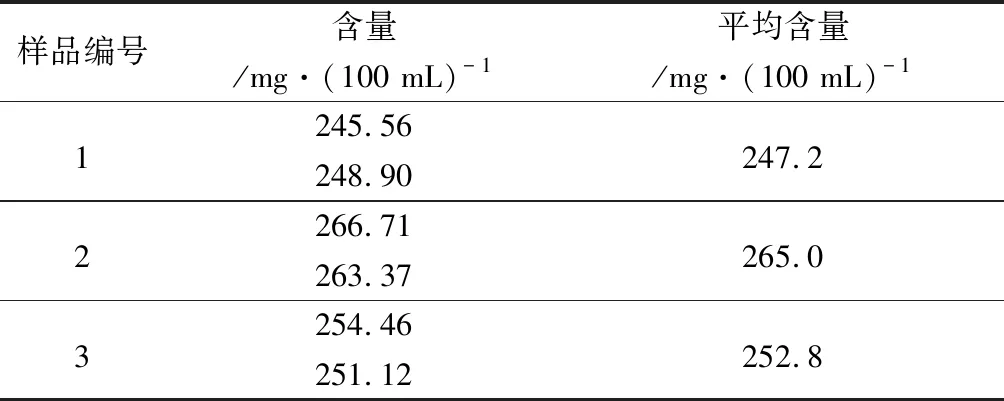
表4 沉淀蛋白質方法樣品測定結果
2.10.2 未沉淀蛋白質方法 按樣品企業標準規定的檢測方法,精密量取3批混合均勻的樣品5 mL,置于100 mL容量瓶中,加水80 mL左右,于沸水浴上加熱2 h,冷卻至室溫后補加水至100 mL刻度,混勻,過濾,棄去初濾液,收集續濾液供沉淀粗多糖。沉淀粗多糖及樣品測定等步驟均同“2.2”項下方法,結果見表5。
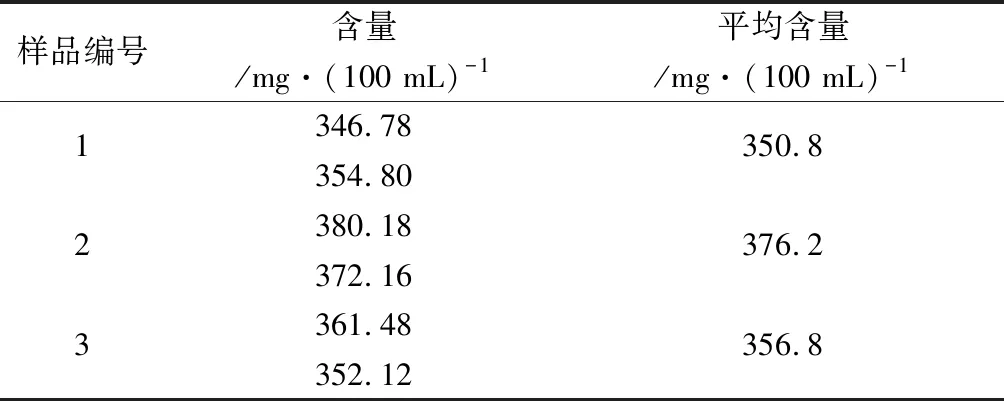
表5 未沉淀蛋白質方法樣品測定結果
由表4~5可知,未沉淀蛋白質方法比沉淀蛋白質方法測得的葡聚糖含量高約106 mg·(100 mL)-1,結果差異較大。為找到結果差異的原因,我們做了以下方面的研究。
2.11 含阿膠陰性空白對照溶液測定及比較 根據企業提供33 kg阿膠制成20 000支口服液(20 mL/支)的配方量,計算得口服液中阿膠濃度為82.5 mg·mL-1。為得到和樣品相同阿膠含量的陰性空白對照,本研究取阿膠,粉碎,過篩,精密稱定8.25 g,置100 mL量瓶中,加水80 mL,水浴煮沸溶解,冷卻,加水,定容至刻度,4 000 r·min-1,離心20 min,取上清液備用。
2.11.1 含阿膠沉淀蛋白質陰性空白溶液的制備 精密量取上述上清液15 mL,按“2.2”項下方法制得樣品測定液。
2.11.2 含阿膠未沉淀蛋白質陰性空白溶液的制備 精密量取上述上清液5 mL,按“2.10.2”項下方法制得樣品測定液。
將上述兩種樣品測定液按“2.2.3”項下方法進行測定,計算葡聚糖含量,結果見表6。

表6 含阿膠陰性空白對照溶液測定結果
由表6可知,阿膠中的蛋白質對粗多糖測定干擾顯著,未沉淀蛋白質的樣品溶液比沉淀蛋白質的樣品溶液測定結果高約91 mg·(100 mL)-1,這與表4~5兩種方法測定結果差值約106 mg·(100 mL)-1相吻合,驗證了本研究采用的沉淀蛋白質前處理方法,能夠有效去除阿膠口服液中蛋白質對粗多糖測定的嚴重干擾。
3 討論
3.1 本研究中阿膠口服液企業標準規定的粗多糖檢測方法有提取步驟,該操作是針對固體樣品的,且長時間沸水浴可能引起糖結構變化,甚至使碳鍵斷裂而導致所測多糖含量偏低。阿膠口服液是均勻澄清的液體,經驗證本法采用沉淀蛋白質的前處理可有效避免上述問題,且能夠有效去除蛋白質帶來的測定干擾。
3.2 比色法測定多糖并非特異性反應,出現測定結果平行偏差較大,所以試驗中沉淀、洗滌、棄上清液、樣液的轉移等操作都要嚴謹,實際操作時可增加平行樣的測定[11]。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
