陽離子型聚丙烯酰胺對含超細氫氧化鎂漿料過濾性能的影響
2020-07-25 07:31:40曹俊雅宋昱霖孫振華李少鵬李會泉
化工進展 2020年7期
曹俊雅,宋昱霖,孫振華,李少鵬,李會泉,3
(1 中國礦業大學(北京)化學與環境工程學院,北京100083;2 中國科學院過程工程研究所綠色過程與工程重點實驗室,濕法冶金清潔生產技術國家工程實驗室,北京100190;3 中國科學院大學化學工程學院,北京100049)
氫氧化鎂作為一種重要的化工產品和無機材料,具有環境友好、無毒、無腐蝕性等優點[1],在阻燃劑、抑煙劑、廢水中和劑、富硫氧化物氣體脫硫劑、染料吸附劑等方面有著廣泛的應用[2-5]。目前制備氫氧化鎂的方法主要是石灰乳沉淀法、氫氧化鈉沉淀法和氨法[6]。但是,采用中和沉淀法制備超細級氫氧化鎂過程中漿料常呈膠體狀,顆粒結晶細小(小于1μm),工業生產過程中沉降和過濾性能較差[7]。
目前提升氫氧化鎂膠體過濾性能的方法通常有晶種法、表面改性法、分散劑法等[8]。余建川等[9]采用在沉鎂過程中加入300%的漿態晶種,發現得到的氫氧化鎂產物的平均粒徑由16μm 增長到43.9μm,過濾時間由平均7min 縮短至50s 左右,晶種法通過加入過量的漿態晶種,調控氫氧化鎂結晶成核過程,增大氫氧化鎂產物的一次粒徑和二次粒徑以提高過濾速率,此方法需要加入大量的漿態晶種,未改變整體漿料的特性,過濾增加效果有限。Yang等[10]利用硅烷偶聯劑對氫氧化鎂固體表面進行改性,發現在氫氧化鎂表面接枝硅烷可以使氫氧化鎂顆粒由2.21μm增大到8.13μm,此方法是增加了固體氫氧化鎂顆粒的粒徑尺寸,與沉淀反應進行中的氫氧化鎂漿料中的氫氧化鎂晶體具有差異,難以直接借鑒使用。Xu等[11]研究了初始試劑濃度、溫度、攪拌速率和添加劑乙二胺四乙酸(EDTA)對氫氧化鎂誘導期的影響,在提前加入少量晶種的前提下加入0.1mol/L EDTA 水溶液能夠改善氫氧化鎂的成核過程,延長誘導期,平均粒徑由原來的12.02μm 增大到20.38μm,但是此過程需要晶種和添加劑共同作用,工藝復雜,對過濾效果增加有限。向蘭等[12]通過在高壓釜內加入特定改性劑溶液在100~250℃之間對常溫合成的氫氧化鎂進行水熱改性,可制備得到高分散片狀氫氧化鎂產品,該方法采用高溫高壓的方式實現氫氧化鎂產品表面性質的改變,提高了氫氧化鎂的分散性能和過濾性能,能耗較高,操作較為復雜。白俊紅等[13]以氯化鎂和氫氧化鈉溶液為原料制備超細氧氧化鎂阻燃劑,利用添加氫氧化鎂理論產量質量的2.63%的硬脂酸時,漿料的濾餅比阻減小了58.27%,該方法通過添加硬脂酸的方式,實現了氫氧化鎂晶體表面性質的改變,提高了過濾分離效率,但是仍存在添加劑用量過大、過濾效果提高有限的缺點。
現有文獻報道均為氫氧化鈉作為沉淀劑制備氫氧化鎂產品的研究,氫氧化鉀作為中和沉淀劑制備氫氧化鎂產品過程的過濾性能缺少系統研究,且簡單有效地改善氫氧化鎂制備過程漿料過濾性能的方法仍需要進一步開發研究,其過濾性能改善機理仍缺少系統分析。同時,采用氫氧化鉀作為沉淀劑從鉀系鹵水或鉀磷肥含鎂溶液中制備氫氧化鎂產品具有重要的應用前景[14],提高超細氫氧化鎂漿料的過濾速率可有效地提高提高氫氧化鎂的生產效率。本文以硫酸鎂溶液、氫氧化鉀為原料,采用前端加入添加劑的方式,完成了不同添加劑對超細氫氧化鎂漿料過濾性能對比篩選,進一步針對最優添加劑進行了單因素試驗的考察,系統研究了添加劑加入方式、添加劑用量比、反應溫度、反應時間和攪拌速率等因素對超細氫氧化鎂漿料過濾速率的影響,采用X 射線衍射儀(XRD)、熱場發射掃描電子顯微鏡(SEM)對氫氧化鎂產物的結構和形貌進行表征,并通過傅里葉紅外光譜儀(FTIR)研究官能團的變化探究陽離子聚丙烯酰胺和氫氧化鎂的結合過程,系統總結了陽離子聚丙烯酰胺改善過濾性能的機理,為超細氫氧化鎂產品的生產過程調控和分離提供了重要的理論指導。
1 實驗材料和方法
1.1 主要儀器和試劑
七水硫酸鎂、硬脂酸,均為分析純,國藥集團化學試劑有限公司;氫氧化鉀,分析純,北京化工廠;聚丙烯酰胺(陽離子型聚丙烯酰胺,分子量1200 萬,根據離子度由低到高編號分別為PAMC01、PAM-C02、PAM-C03;陰離子型聚丙烯酰胺,型號PAM-A01 分子量為1200 萬,型號PAMA02 分子量為1600 萬,型號PAM-A03 分子量為1800 萬;非離子型聚丙烯酰胺,PAM-N01,分子量為1000萬),北京海暢清環保科技有限公司;油酸,分析純,上海阿拉丁生化科技股份有限公司;聚乙二醇(PEG,PEG 6000 為分子量6000,PEG 20000為分子量20000),國藥集團化學試劑有限公司,均為化學純;實驗用水均為超純水。
電感耦合等離子體發射光譜儀(ICP-OES,Avio200 型),PerkinElmer 公司;三孔恒溫水浴鍋(HH-3D 型),北京隆盛創新商貿中心;數字黏度計(LVDV-2 型),上海精天電子儀器有限公司;循環水式多用真空泵(SHZ-95B 型);蘭格分配型蠕動泵(BT100-1F),保定蘭格恒流泵有限公司;熱場發射掃描電子顯微鏡(SEM,JSM-7610F型),日本電子株式會社北京事務所;X 射線衍射儀(XRD,Empyrean型),荷蘭帕納科公司;傅里葉紅外光譜儀(FTIR,Tensor27型),德國布魯克公司。
1.2 實驗方法
實驗裝置見圖1。首先將七水硫酸鎂配制成1L硫酸鎂溶液[c(Mg2+)=10g/L],取200mL 于三口燒瓶中,放入恒溫水浴鍋中,調節攪拌頭轉速,待溫度和轉速穩定后通過蠕動泵以1.45mL/min 的滴加速率向三口燒瓶中加入質量分數為30%的氫氧化鉀溶液,加料完畢后繼續攪拌反應2.5h,進行不同條件下的單因素實驗。反應結束后利用黏度計進行黏度測試,趁熱進行抽濾分離、洗滌,記錄抽濾時間和濾液體積,并通過式(1)[15]計算過濾速率。最后將濾餅放到100℃烘箱中干燥備用。

式中,ν 為過濾速率,mL/(m2·s);V 為濾液體積,mL;s為濾餅面積,m2;t為過濾時間,s。

圖1 實驗裝置
采用SEM 觀測產品的形貌特征;采用ICPOES 測試液相中Mg2+的濃度;采用XRD 對產物進行晶體結構分析;采用FTIR 觀測官能團的變化來探究產物與陽離子聚丙烯酰胺的絮凝劑機理。
單因素考察條件:添加劑種類(陽離子聚丙烯酰胺、硬脂酸、油酸、PEG)、反應溫度(40℃、60℃、70℃、80℃)、攪拌速率(150r/min、200r/min、250r/min、300r/min、350r/min、400r/min)、添加劑用量比ε(ε 為陽離子聚丙烯酰胺與理論生成氫氧化鎂的質量分數,其中ε=0、0.1%、0.3%、0.5%、0.7%、0.9%、1%、1.2%、1.4%)、添加劑的添加方式等因素。
2 結果與討論
2.1 添加劑種類對氫氧化鎂漿料過濾速率的影響
圖2為60℃、300r/min、ε=2%下不同種類添加劑對氫氧化鎂膠體過濾速率的影響。由圖可見,硬脂 酸、PEG6000、PEG20000、PAM-A01、PAMN01、PAM-C01對超細氫氧化鎂漿料過濾效果起抑制作用,油酸添加對超細氫氧化鎂漿料過濾性能影響 不 大,而PAM-A02、PAM-A03、PAM-C02 和PAM-C03 這4 種添加劑對膠體過濾速率具有明顯的促進作用,其中PAM-C03 型的影響最為顯著,過濾速率為184mL/(m2·s)。因此,后續的實驗條件采用PAM-C03為最優添加劑。
2.2 添加方式對氫氧化鎂漿料過濾速率的影響

圖2 添加劑的種類對氫氧化鎂漿料過濾速率的影響

圖3 PAM-C03添加方式對氫氧化鎂漿料過濾速率的影響
圖3為在60℃、250r/min下不同濃度PAM-C03的不同添加方式對超細氫氧化鎂漿料過濾速率的影響。通常采用PAM-C03 配制成水溶液進行實驗,而與采用直接加入PAM-C03 的方法對比可以看出在相同濃度下直接加入法的過濾速率略優于配制成水溶液的過濾速率,并且隨著添加量的增加而增大。另外,采用直接加入法流程簡單、過程中用水量少。因此,采用直接添加PAM-C03 能夠有效地加快氫氧化鎂漿料的過濾速度。
2.3 添加劑用量比對氫氧化鎂漿料過濾速率的影響
圖4 為在60℃、250r/min 下PAM-C03 濃度對超細氫氧化鎂漿料過濾速率的影響。由圖可見,在ε<0.7%時,隨著PAM-C03 濃度的增加,超細氫氧化鎂漿料的過濾速率逐漸增大;當ε>0.7%時,超細氫氧化鎂漿料的過濾速率呈現逐漸下降的趨勢;其中在ε=0.7%處時,氫氧化鎂漿料的過濾速率最大為545mL/(m2·s)。因此,選用ε=0.7%為最優濃度。

圖4 添加劑用量比對氫氧化鎂漿料過濾速率的影響
2.4 反應溫度對氫氧化鎂漿料過濾速率的影響
圖5為在ε=0.7%、250r/min下不同反應溫度對超細氫氧化鎂漿料過濾速率的影響。由圖可見,含超細氫氧化鎂漿料的過濾速率隨著反應溫度的升高呈先上升后下降的趨勢,當反應溫度為60℃時,漿料的過濾速度最快,達到545mL/(m2·s)。隨著溫度從40℃上升到60℃的過程中,含氫氧化鎂漿料黏度逐漸變小,體系布朗運動加快,PAM-C03 與溶液中生成的氫氧化鎂顆粒的碰撞次數增多,因此絮凝過程逐漸加快。隨著溫度從60℃升高到80℃,漿料的過濾速率降低,原因是由于溫度的升高容易使PAM-C03 老化甚至分解為不溶物質,從而降低絮凝過程,使漿料的過濾速率下降[16]。因此,選用60℃為最優反應溫度。

圖5 反應溫度對氫氧化鎂漿料過濾速率的影響
2.5 攪拌速度對氫氧化鎂漿料過濾速率的影響
圖6為在60℃、ε=0.7%下攪拌速度對超細氫氧化鎂漿料過濾速率的影響。由圖可見,隨著攪拌速度從150r/min增加至250r/min時,含超細氫氧化鎂漿料的過濾速率從231mL/(m2·s)增長到545mL/(m2·s)。說明在反應初期隨著攪拌速率的增加,PAM-C03與溶液中生成的氫氧化鎂顆粒的碰撞機會越來越大,分子鏈上吸附架橋能力也越來越強。但隨著攪拌速率繼續增大,漿料的過濾速率逐漸下降,說明攪拌速率過快可能會使PAM-C03 與氫氧化鎂絮體結合形成的大絮體被破壞,從而使過濾速率減慢。因此,選用250r/min為反應的最優攪拌速率。
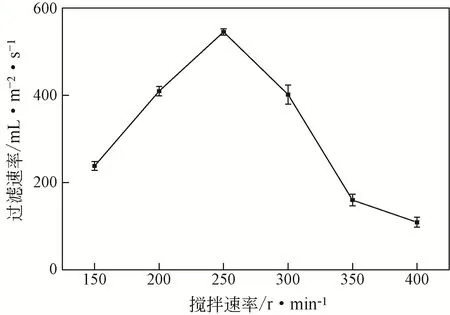
圖6 攪拌速度對氫氧化鎂漿料過濾速率的影響
2.6 添加劑用量比對氫氧化鎂漿料黏度的影響
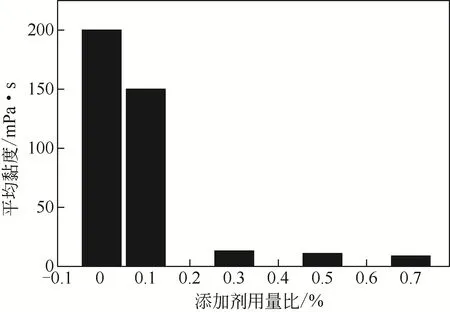
圖7 添加劑用量比對氫氧化鎂漿料黏度的影響
圖7 為在60℃、250r/min 下添加劑用量比對含氫氧化鎂漿料黏度的影響。由圖可見,隨著PAM-C03添加劑用量比的增加,體系的黏度值呈現逐漸降低的趨勢,當添加劑用量比大于0.3%時,漿料黏度降低幅度減弱,不添加PAM-C03 時,漿料黏度為200mPa·s,添加劑用量比為0.7%時漿料黏度下降到8.64mPa·s,僅為不添加添加劑黏度值的4.32%。說明PAM-C03的加入能夠改變顆粒間的連接方式,從而降低黏度增加了漿料的流動性,使得含氫氧化鎂漿料的過濾速率加快。
2.7 添加劑用量比對反應過程中氫氧化鎂晶體形貌的影響
不同添加劑用量比下所得氫氧化鎂晶體的SEM 圖見圖8。從圖8(a)可見,不添加PAM-C03時,氫氧化鎂晶體小顆粒密實的結合、顆粒之間團聚嚴重,晶體平均粒徑較小,平均粒徑(D50)=89nm;隨著添加劑用量比增加,顆粒粒徑呈逐漸增大的趨勢,達到D50=125nm,顆粒之間結合團聚降低。其中由圖8(c)、(d)可知,添加劑用量比為0.5%、0.7%時氫氧化鎂顆粒粒徑明顯增大,D50=138nm,晶體呈六方片狀形貌;由圖8(e)、(f)可知,隨著PAMC03用量比進一步增加,氫氧化鎂晶體顆粒之間團聚現象重新增強,分散程度降低,D50=81nm,這說明PAM-C03 添加過量時,聚丙烯酰胺分子與水發生凝膠化過程,增加了體系的黏度,晶體之間結合降低,這與過濾速率結果和黏度數值結果一致。
2.8 不同添加劑用量比下氫氧化鎂結合機理分析
圖9為不同添加劑用量比下所得氫氧化鎂晶體的XRD譜圖。由圖可見,不添加PAM-C03和添加PAM-C03 得到的氫氧化鎂的特征峰與純氫氧化鎂標準譜圖對應,由此可以證明加PAM-C03 后得到的產物是氫氧化鎂。其特征峰對應的晶面依次為(001)、(011)、(012)、(110)、(111)、(103)、(201)和(202)。隨添加劑用量比的增加,氫氧化鎂產物對應的(011)晶面的半峰寬呈現先下降后升高的趨勢,說明添加劑用量比由0 增至0.7%時氫氧化鎂的晶粒增大,但隨著添加劑用量比的繼續增加,晶粒減小。
表1為不同添加劑用量比下(001)面和(011)面對應的衍射峰的相對強度比值。由表1可知,隨著添加劑用量比增加,氫氧化鎂特征衍射峰的相對強度發生變化。添加劑用量比由0.1%增至0.7%時,(001)面和(011)面對應的衍射峰相對強度逐漸減小,結晶度變差。繼續增加添加劑用量比,衍射峰的相對強度稍有增加后又減小,說明PAMC03的加入使氫氧化鎂產物的結晶性能變差,這與SEM結果相吻合。

圖8 不同添加劑用量比下所得氫氧化鎂晶體的SEM圖(插圖為粒徑分布圖)

圖9 不同添加劑用量比下氫氧化鎂晶體的XRD譜圖

表1 不同添加劑用量比下(001)面和(011)面對應的衍射峰的相對強度比值
圖10(a)為有無PAM-C03 下氫氧化鎂產物的FTIR譜圖。由圖可見,PAM-C03在3446cm-1處為酰胺基中的反對稱伸縮振動吸收峰,2921cm-1處為甲基和亞甲基的吸收峰,2860cm-1處為的振動吸收峰,1756cm-1處出現了酰氧基團的特征吸收峰,并且1653cm-1處出現了酰胺羰基的C==O伸縮振動吸收峰,1111cm-1處為 N+(CH3)3上甲基的特征吸收峰。添加劑用量比為0.7%時所制氫氧化鎂晶體的FTIR譜圖上出現了PAM-C03的酰胺氧基團、酰胺羰基以及官能團所帶甲基亞甲基對應的特征峰,說明PAM-C03 的加入,使氫氧化鎂晶體上的羥基官能團與PAM-C03的官能團相結合。

圖10 不同添加劑用量比下氫氧化鎂晶體的FTIR譜圖
圖10(b)為不同添加劑用量比下氫氧化鎂產物的FTIR 譜圖。通過對比能夠看出,3698cm-1處為氫氧化鎂的特征峰,不同添加劑用量比下產物的不同位置上出現了PAM-C03 的特征峰,3445cm-1處出現了特征吸收峰,并且隨著添加劑用量比的增加,吸收程度越明顯;1118cm-1處出現了的特征吸收峰,說明加入PAM-C03后不同添加劑用量比下生成的氫氧化鎂中的結合了部分PAM-C03 中的酰胺基團,且添加劑用量比越高,特征吸收峰強度越明顯。

圖11 氫氧化鎂與PAM-C03的結合過程
圖11為氫氧化鎂與PAM-C03的結合過程,其中PAM-C03 中酰胺羰基上的π 電子與氮原子上未共用電子對形成P-π共軛體系[17],使氮原子上電子云密度降低,而羰基上氧原子的電子云密度升高,更容易得到質子,形成更強的氫鍵。PAM-C03 的分子鏈上含有大量的酰胺基團,能與氫氧化鎂的一個或多個羥基形成氫鍵[18],因此PAM-C03 能夠有效地吸附架橋到氫氧化鎂顆粒表面,這也證明了在FTIR譜圖中出現的在加入PAM-C03后生成的氫氧化鎂中出現了PAM-C03 的特征官能團,并且形成了PAM改性Mg(OH)2的結構。由于氫氧化鎂顆粒之間靜電斥力較強,分子間極易團聚,親水性強,極易形成網狀結構導致沉降過濾性能差,PAM-C03的加入使兩者相互連接形成大分子長鏈,產生架橋效應,從而阻止顆粒間的接觸,形成粗大的絮團[19],降低了體系液相的黏度,加快了體系內液體的流動性,因此含氫氧化鎂漿料的過濾速率顯著提高。
3 結論
(1)采用硫酸鎂、氫氧化鉀為原料探究出了氫氧化鎂漿料過濾速率最優的工藝條件:陽離子型聚丙烯酰胺PAM-C03、添加方式為直接加入法、反應溫度60℃、攪拌速率250r/min、添加劑用量比為0.7%。
(2)最優條件下氫氧化鎂漿料的過濾速率由不添加聚丙烯酰胺時的73mL/(m2·s)提升到545mL /(m2·s),體系中漿料的黏度由200mPa·s下降到8.64mPa·s。表明PAM-C03 的存在能夠有效地減小膠體溶液的黏度,提高超細氫氧化鎂漿料的過濾速率。
(3)PAM-C03的加入不改變氫氧化鎂的晶體結構,其提高過濾速率的機理主要是使氫氧化鎂顆粒的聚集狀態發生改變,PAM-C03分子鏈上的酰胺基團能吸附架橋到氫氧化鎂晶體表面,與氫氧化鎂晶體中的羥基相連形成長鏈,減少了氫氧化鎂顆粒間的團聚,有效地提高了超細氫氧化鎂漿料的過濾速率。
