金銀花總有機酸有效部位的制備及質量標準研究
2020-07-23 00:50:52王騰騰蘇培文李孟孟李海剛
亞太傳統醫藥 2020年6期
張 蕊,王騰騰,蘇培文,李孟孟,黃 鵬,李海剛
(臨沂大學,山東 臨沂 276000)
有機酸類化合物是金銀花的主要有效成分[1-2],包括綠原酸、異綠原酸、咖啡酸等有機酸類成分,具有抑菌抗病毒[3-4]、解熱[5]、抗炎、保肝、止血、抗氧化、免疫調節等作用[6]。近年來人們對金銀花綠原酸的提取方法[7-8]、金銀花有機酸類成分的含量測定方法進行了廣泛的研究,但將金銀花總有機酸作為有效部位進行分離提取的報道尚不多見。本文在前期研究的基礎上制備了金銀花總有機酸有效部位,研究其薄層鑒別方法、總有機酸含量測定方法,采用高效液相色譜法測定了金銀花總有機酸有效部位中綠原酸、異綠原酸A、異綠原酸B及異綠原酸C的含量,為金銀花總有機酸有效部位的質量標準研究提供依據,為金銀花總有機酸有效部位制劑的開發提供參考。
1 儀器與試藥
1.1 儀器
1260型高效液相色譜儀(美國 Agilent公司);TU-1810spc紫外可見分光光度計(北京普析通用儀器有限公司);旋轉蒸發儀(RE-2000A上海亞榮生化儀器廠);ME104E電子分析天平(梅特勒-托利多儀器有限公司)。
1.2 試藥
綠原酸對照品(上海源葉生物科技有限公司,批號:Y24J7K16726);異綠原酸A對照品(中國食品藥品檢定研究院,批號:P28O7F23862);異綠原酸B對照品(中國食品藥品檢定研究院,批號P25J6F1793);異綠原酸C對照品(中國食品藥品檢定研究院,批號:P28D4S1);金銀花(山東平邑);甲醇、乙腈為色譜純,水為純凈水,其余試劑均為分析純。
2 方法與結果
2.1 金銀花總有機酸有效部位的制備
取金銀花切段,加入10倍量70%乙醇回流煎煮3次,每次1h,濾過,合并3次煎煮液,濾過,取續濾液減壓濃縮至相對密度1.2,石油醚萃取除去脂溶性成分,然后用等量乙酸乙酯萃取5次,棄去乙酸乙酯層,取水層上大孔吸附樹脂LSA-7,徑高比9∶1,先用4倍量柱體積水洗脫,洗脫流速3 mL/min,再用7倍量70%乙醇洗脫,洗脫流速5 mL/min,收集乙醇洗脫液,減壓濃縮至干,即得金銀花總有機酸有效部位,其中總有機酸含量約為71.6%。
2.2 薄層色譜鑒別
取金銀花有機酸有效部位粉末50 mg置于10 mL量瓶中,加75%乙醇使溶解,作為供試品溶液。量取綠原酸對照品,加75%乙醇制成每1 mL含1 mg的溶液。照薄層色譜法(中國藥典2015年版通則0502)試驗,分別吸取上述兩種溶液各10 μL,點于同一硅膠G薄層板上,以乙酸乙酯∶水∶乙酸∶二甲苯(85∶10∶20∶4)溶液為展開劑,飽和20 min后展開,取出,晾干,于365nm波長處觀察,供試品色譜圖中,在與對照品色譜相應的位置上,顯相同顏色的斑點。結果見圖1。
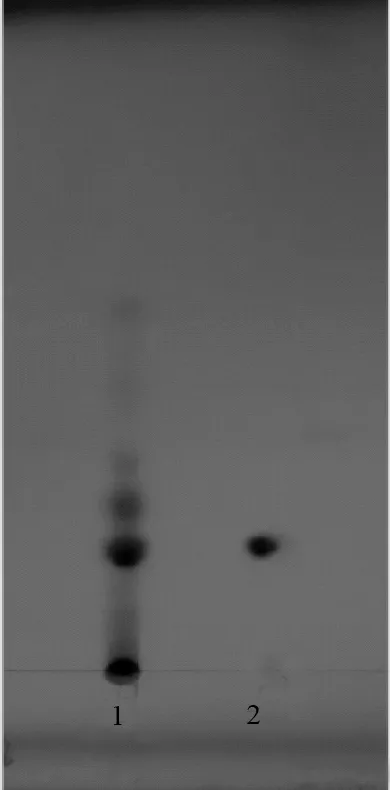
1.金銀花有機酸有效部位;2.綠原酸
2.3 總有機酸含量測定
2.3.1 綠原酸對照品溶液配制 精密稱取綠原酸對照品0.010 8 g,置于50 mL棕色量瓶中,加70%乙醇溶解并定容至刻度,搖勻,即得質量濃度為0.216 mg/mL的對照品溶液(10 ℃以下避光保存)。
2.3.2 最大吸收波長選擇 取綠原酸對照品溶液適量,加70%乙醇稀釋至適當濃度,按照紫外分光光度法于200~600 nm波長范圍內進行紫外掃描,記錄掃描色譜圖,結果如圖2所示。取金銀花總有機酸提取物適量,70%無水乙醇溶解并稀釋至適當濃度,同法于200~600 nm波長處掃描,記錄掃描色譜圖,結果如圖3所示。
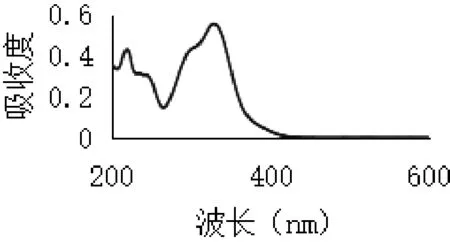
圖2 綠原酸紫外掃描
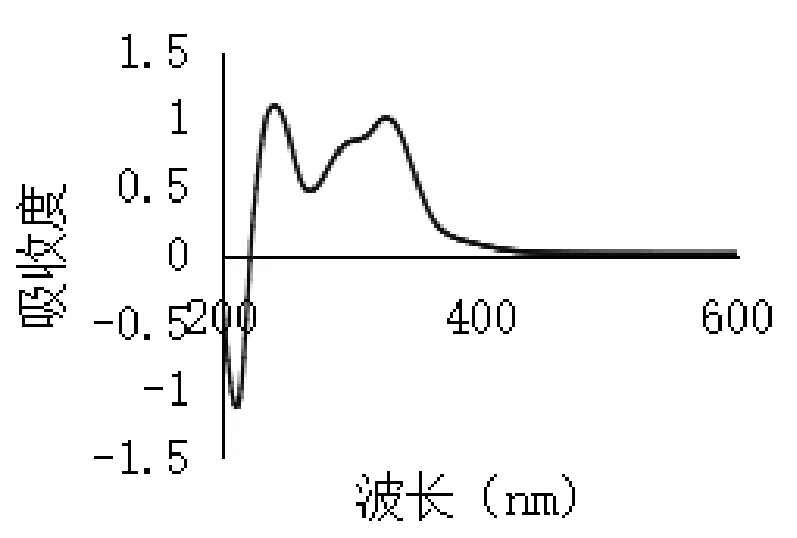
圖3 總有機酸有效部位紫外掃描
由圖可見,綠原酸在327 nm波長處有一最大吸收,總有機酸在243 nm及327 nm波長處有最大吸收,參考山東省藥材標準[9](金銀花提取物中總有機酸測定方法),結合本實驗結果,本文選擇327 nm作為吸收波長測定總有機酸含量。
2.3.3 標準曲線制備 精密量取綠原酸對照品溶液0.2 mL、0.4 mL、0.6 mL、0.8 mL、1.0 mL,置于10 mL容量瓶中,用70%乙醇稀釋至刻度,以70%乙醇作空白對照,在327 nm處測吸光度。以質量濃度(x)為橫坐標,吸光度(y)為縱坐標,繪制標準曲線,綠原酸在4.32~21.6 μg/mL濃度范圍內與其吸光度線性關系良好,回歸方程為y=48.936x+0.0425(R2=0.9994)。
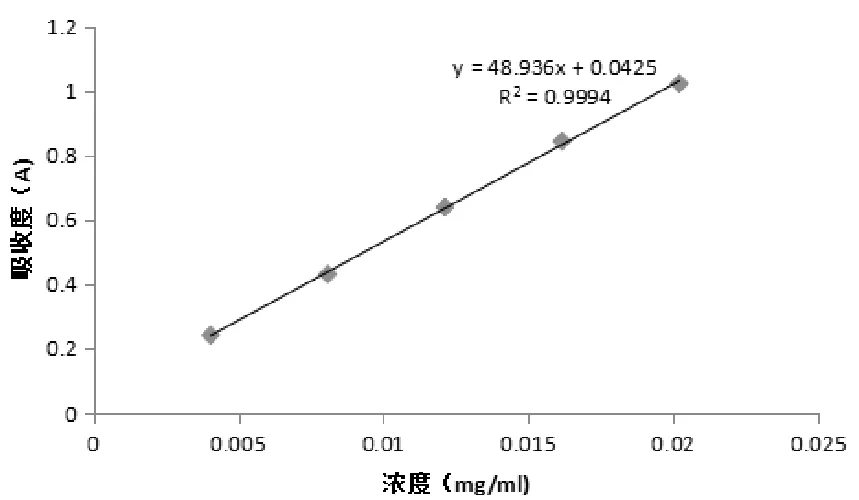
圖4 綠原酸標準曲線
2.3.4 穩定性試驗 精密量取綠原酸對照品溶液0.6 mL置于10 mL棕色容量瓶中,用70%乙醇稀釋至刻度,搖勻,在0、2、4、6、8 h,于327 nm下測定吸光度。RSD為0.98%,溶液吸光度在8h內基本穩定。
2.3.5 精密度試驗 精密量取綠原酸對照品溶液0.6 mL置于10 mL棕色容量瓶中,用70%乙醇稀釋至刻度,搖勻,于327 nm下測定吸光度,連續測定6次,RSD為0.56%,表明試驗所用儀器精密度良好。
2.3.6 樣品回收率試驗 分別精密量取綠原酸對照品溶液0.6 mL 9份,分別置于9個25 mL量瓶中,各量瓶均加入已知濃度的供試品溶液0.4、0.5、0.6 mL,70%乙醇稀釋至刻度,搖勻,于327 nm下測定吸光度,計算總有機酸加樣回收率,結果見表1,RSD為1.93%,說明該方法準確度良好。
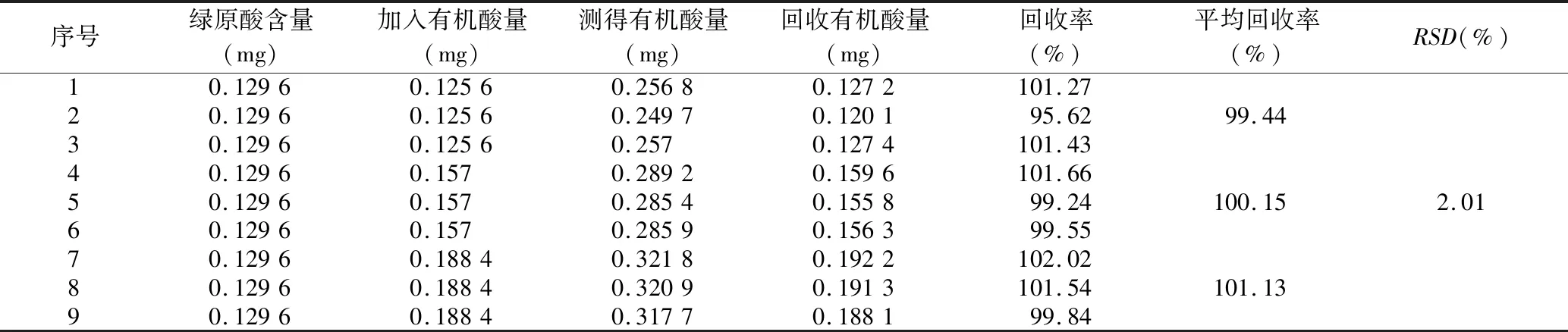
表1 總有機酸的加樣回收率試驗結果
參考山東省中藥材標準中“金銀花提取物”總綠原酸含量測定方法[9],結合本實驗做如下變動,測定樣品中總綠原酸含量,具體測定方法如下:精密稱取綠原酸對照品0.010 8 g,置于50 mL棕色量瓶中,加70%乙醇溶解并定容至刻度,搖勻,即得質量濃度為0.216 mg/mL的對照品溶液(10 ℃以下避光保存)。取提取物粉末置于25 mL棕色量瓶中,加70%乙醇超聲溶解并稀釋至刻度,搖勻、濾過得供試品溶液。按照紫外分光光度法于327 nm波長處測定吸收度,以綠原酸為對照計算總有機酸含量,結果見表2。
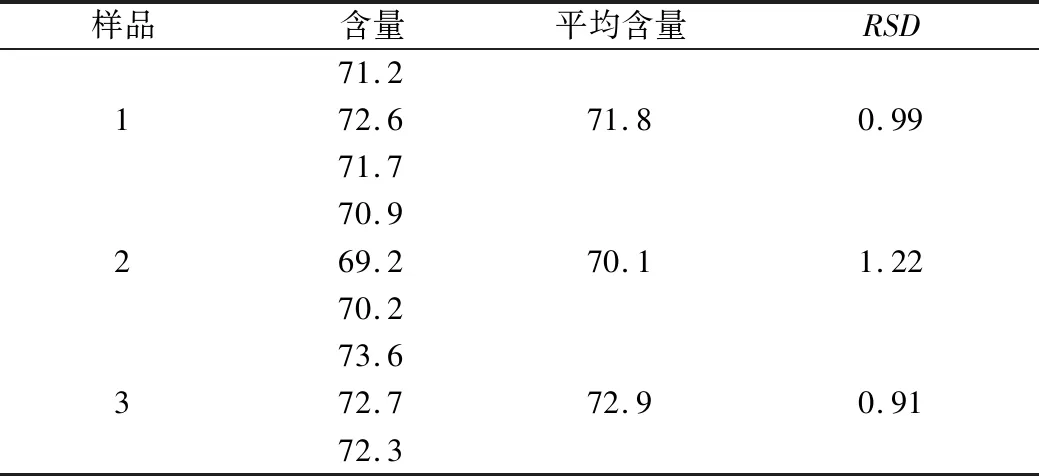
表2 金銀花有機酸有效部位中總有機酸含量測定結果(%)
2.4 樣品中指標成分含量測定
以綠原酸、異綠原酸A、異綠原酸B、異綠原酸C作為指標性成分,采用高效液相色譜法測定各指標性成分含量。
2.4.1 色譜條件 以十八烷基硅烷鍵合硅膠為填充劑;以乙腈為流動相A,以0.4%磷酸溶液為流動相B,梯度洗脫:0~15 min,10%~20%(A);15~40 min,20%~30%(A);40~60 min,30%~60%(A);檢測波長為327 nm;柱溫:室溫;流速為1 mL·min-1;進樣量為10 μL。
2.4.2 混合標準品溶液配制 分別精密稱取綠原酸0.010 1 g、異綠原酸A 0.010 0 g、異綠原酸B 0.010 5 g、異綠原酸C 0.010 1 g,分別置于10 mL量瓶中,加50%甲醇溶解并稀釋至刻度,得各指標性成分儲備液。分別取各指標性成分儲備液2.5 mL置于25 mL量瓶中,加50%甲醇溶解并稀釋至刻度,得各指標性成分混合標準品溶液。
2.4.3 系統適應性 取混合標準品溶液,按照“2.4.1”項下色譜條件測定,綠原酸、異綠原酸B、異綠原酸A、異綠原酸C分離度良好,相鄰色譜峰分離度均大于1.5,理論塔板數按綠原酸計算不低于2 000。
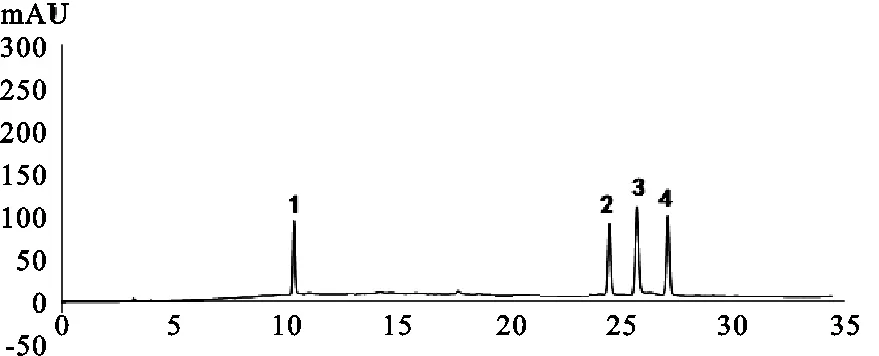
1.綠原酸;2.異綠原酸B;3.異綠原酸A;4.異綠原酸C
2.4.4 標準曲線制備 分別精密吸取“2.4.2”項下混合標準品溶液0.1、1.0、3.0、5.0 mL,置于10 mL量瓶中,70%乙醇稀釋至刻度,搖勻,得系列濃度的混合標準品溶液,分別吸取10 μL注入液相色譜儀,記錄色譜圖,以峰面積對濃度進行線性回歸,得回歸方程,各對照品回歸方程見表3。可見4種成分在0.01~1.0 mg·mL-1濃度范圍內與其峰面積積分線性關系良好。
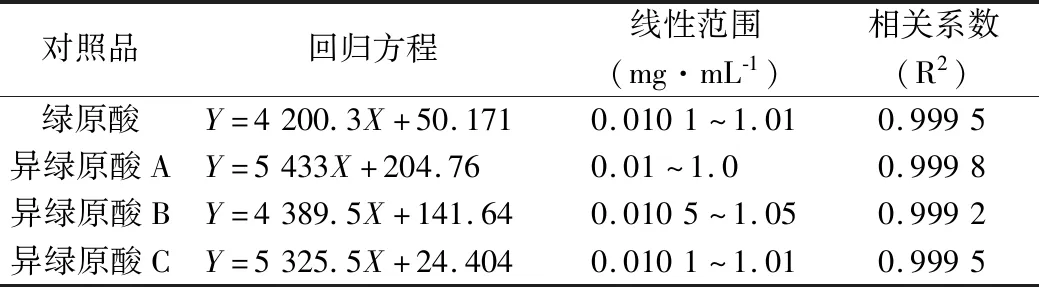
表3 4種對照品的回歸分析結果
2.4.5 精密度試驗 精密吸取混合對照溶液1.0 mL置于10 mL量瓶中,加70%的乙醇稀釋至刻度,搖勻,分別取10 μL連續進樣6次,結果綠原酸、異綠原酸A、異綠原酸B、異綠原酸C峰面積RSD分別為0.62%、0.77%、0.82%。
2.4.6 穩定性試驗 精密吸取混合對照溶液1.0 mL置于10 mL量瓶中,加70%的乙醇稀釋至刻度,搖勻,分別于0、2、4、6、8、12 h取10 μL進樣分析,結果綠原酸、異綠原酸A、異綠原酸B、異綠原酸C峰面積RSD分別為0.72%、0.85%、0.94%。表明在12 h以內各對照品溶液穩定性良好。
2.4.7 重復性試驗 精密吸取混合對照溶液1.0 mL 6份,分別置于10 mL量瓶中,加70%的乙醇稀釋至刻度,搖勻,分別取10 μL進樣分析,結果綠原酸、異綠原酸A、異綠原酸B、異綠原酸C峰面積RSD分別為0.51%、0.65%、0.74%,表明重復性良好。
2.4.8 加樣回收率試驗 分別精密稱取已知含量的同一樣品3份若干置于100mL量瓶中,分別精密加入各對照品溶液適量,加70%的乙醇稀釋至刻度,搖勻,精密稱取10 μL注入高效液相色譜儀,計算3種成分的加樣回收率。由表4結果可以看出,在該色譜條件下,該定量方法對3種成分的加樣回收率良好。
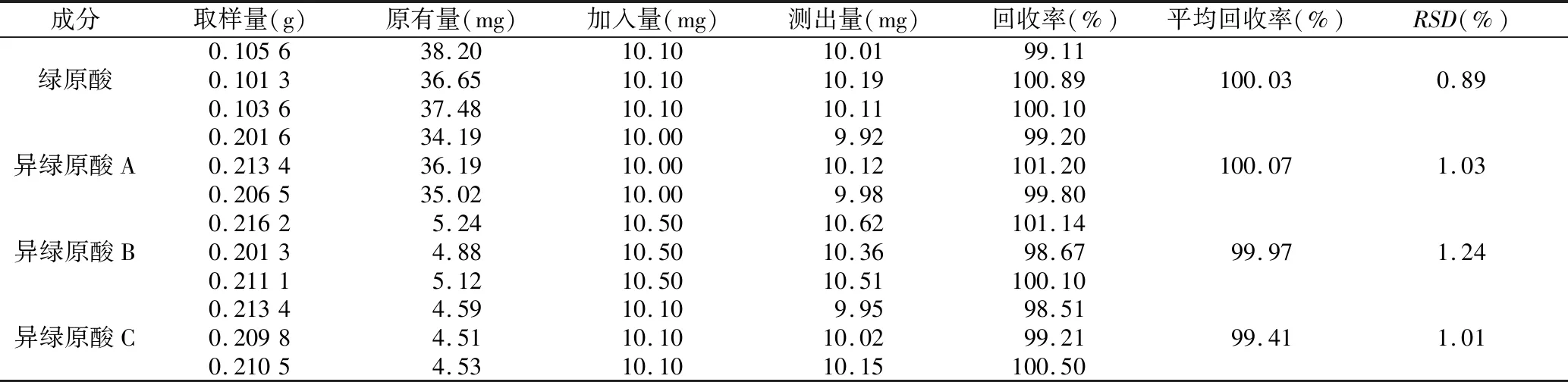
表4 4種成分的加樣回收率試驗結果
2.4.9 樣品中綠原酸、異綠原酸B、異綠原酸A、異綠原酸C含量測定 取有機酸提取物樣品置于100 mL量瓶中,加70%乙醇超聲溶解并稀釋至刻度,取10 μL注入液相色譜儀,記錄色譜,按外標法計算樣品中4種成分含量。見表5。

表5 金銀花有機酸有效部位中4種成分含量測定結果
3 討論
有機酸有效部位薄層鑒別圖譜中綠原酸斑點清晰,其他成分斑點不夠清晰,可能是含量較低的原因,因此在薄層鑒別中我們選擇綠原酸作為有機酸有效部位薄層鑒別指標性成分。
流動相篩選過程中,考察了乙腈-水、甲醇-水、乙腈-磷酸、甲醇-磷酸、乙腈-醋酸、甲醇-醋酸梯度洗脫,發現以乙腈-磷酸為流動相時色譜峰峰形較好,分離度較高。綠原酸對照品溶液在200~800 nm全波長掃描,結果表明綠原酸在327 nm波長處有最大吸收。總有機酸提取物的200~600 nm全波長掃描結果顯示出兩個吸收峰(327 nm、243 nm),參照山東省中藥材標準中“金銀花提取物”總綠原酸含量測定方法測定樣品中總綠原酸含量,選擇327 nm作為檢測波長。
