電化學提鋰技術中電極材料和電極體系的研究進展
2020-06-29 04:06:02郭志遠紀志永陳華艷時雅濱張帆趙穎穎劉杰袁俊生
化工進展 2020年6期
關鍵詞:體系
郭志遠,紀志永,陳華艷,時雅濱,張帆,趙穎穎,劉杰,袁俊生
(1 河北工業大學化工學院/化工節能過程集成與資源利用國家-地方聯合工程實驗室,天津300130;2 河北工業大學化工學院/海水資源高效利用化工技術教育部工程研究中心,天津300130;3 河北省現代海洋化工技術協同創新中心,天津300130;4 天津工業大學分離膜與膜過程國家重點實驗室,天津300387;5 北京交通大學海濱學院,河北黃驊061199)
鋰及其化合物被廣泛應用于玻璃、陶瓷、電子、新能源汽車、軍工國防和航空航天等領域。近年來,隨著電子產品和新能源汽車的快速發展,使得對鋰離子電池的需求量與日俱增[1-2]。根據相關全球鋰資源供需分析,2023 年鋰資源供應將不能滿足需求,因此鋰資源的開發成為研究熱點[3]。
當前,鋰資源的主要來源為鋰礦石和鹽湖鹵水。由于礦石提鋰存在高能耗和高污染的缺點,加之鹽湖鹵水占陸上總量的70%~80%,鹽湖鹵水提鋰已成為獲取鋰資源的主要途徑。隨著富鋰鹽湖鹵水資源的快速開發利用,開發對象逐漸轉向低品位鹽湖鹵水鋰資源[4]。與此同時,伴隨大規模海水淡化產業的蓬勃發展,減少濃海水的直排,并進行資源化利用(海水中鋰素儲量達2600 億噸,為陸上總量的萬余倍)成為共識[5]。因此,低品位鹽湖鹵水/(濃)海水成為下一階段獲取鋰資源的重要渠道。由于低品位鹽湖鹵水(Li+濃度<24.5mg/L)和(濃)海水(0.17~0.34mg/L)等溶液中鋰的相對含量低,且共存多種堿金屬和堿土金屬離子(如K+、Na+、Mg2+、Ca2+),其中,與鋰離子物性相近的Mg2+含量較高(國內鹽湖鹵水中Mg/Li 質量比一般在35 以上,(濃)海水中的比值則高達7400 多),使得其開發更為困難。因此,鋰的選擇性提取和高效富集成為低品位鋰資源提取技術需求的核心[6]。傳統的蒸發沉淀法僅適用于Li+濃度較高、低鎂鋰比的鹵水。針對這些復雜稀薄溶存鋰資源的開發利用,納濾[7]、電滲析[8-9]、離子交換與吸附法[10-11]、電化學提鋰法逐漸被開發。其中,電化學提鋰法具備選擇性高、適用于低品位鋰資源、環境友好的優勢而受到廣泛關注。
在電化學提鋰過程中,電極材料的選擇/制備和提鋰電極結構體系/裝備的構建是影響電化學提鋰過程的兩個最主要因素。本文圍繞近年來國內外電化學提鋰技術的研究,對鋰吸附電極材料的選擇/制備和提鋰電極結構體系的發展兩方面進行了總結分析,并針對目前電化學提鋰過程存在的問題及不足指出后續研究方向,以期為電化學提鋰技術更好的發展和應用提供思路和參考。
1 電化學技術原理
電化學提鋰是利用鋰離子電池(lithium ion batteries,LIBs)正極材料在充放電過程中會伴隨Li+在固相電極和液相電解液兩相之間轉移的原理來實現的。在充電過程中正極材料被氧化,Li+從正極材料中脫出到電解液中;在放電過程中,正極材料被還原,Li+從電解液中嵌入到正極材料中[12-13]。基于放電過程,把LIBs里的有機電解液換為含鋰的鹵水/濃海水,可實現Li+的提取;基于充電過程,把LIBs 里的有機電解液換為成分單一的回收液,即可實現Li+的回收。
電池的正極材料主要有LiFePO4、LiMn2O4和LiCoxMnyNi1-x+yO2等[14]。其中,尖晶石型LiMn2O4因其高的Li+選擇性,作為離子篩(能吸附特定離子的復合氧化物多孔材料)用在鋰資源的提取上已得到廣泛應用[15-16],其應用包括吸附(吸附目標離子至離子篩內)和洗脫(用酸性或強氧化性洗脫劑將目標離子從離子篩內再生出來)兩個過程。對于洗脫過程,常用的洗脫劑為HCl、H2SO4和HNO3等,后期為抑制酸洗過程中Mn的溶損,過硫酸鹽洗脫劑逐漸被開發應用[15,17]。當前,離子篩對于Li+的吸附機理被認為是由離子交換與吸附和氧化還原兩類反應共同作用的[18-19]。
電化學提鋰避免了離子篩再生過程中酸性或強氧化性洗脫劑的使用,不僅實現了提鋰的環境友好性,還避免了洗脫劑對離子篩結構的破壞,具有適用于低品位鹵水、環境友好、能耗低等優勢。電化學提鋰的成本主要為設備(電極材料及裝置)成本和運行成本。在運行成本上,由于電極材料對Li+的高選擇性,在Li+的分離純化和富集過程中其僅消耗少量電能(≤30W·h/mol Li+),低于電滲析的100~2000W·h/mol Li+[9]。因此,通過電極材料和裝置優化控制其設備成本后,電化學提鋰技術將具有廣泛的應用前景。電極材料的制備和電極體系構建的參考條件分別為:Li+交換容量、選擇性、循環穩定性等;電極體系的設備成本、提鋰過程能耗和提鋰效率等。
2 Li+脫/嵌電極材料
在電化學提鋰過程中,一般存在兩個電極。其中,“工作電極”用來實現Li+的選擇性嵌入/脫出過程;“對電極”用來與工作電極形成閉合回路,其主要分為電子導體和離子導體兩類,以使得電化學提鋰體系保持電中性。此外,在一些研究中還會加入Ag/AgCl 或飽和甘汞電極做參比電極,并用來穩定電極電勢。通常,工作電極材料與鋰離子電池的正極材料相似,選擇的主要參考條件為電極的活性材料對鋰的選擇性和交換容量及其循環穩定性等。工作電極的活性材料對于Li+的高效選擇性主要由其晶格空位大小所決定。Li+在所有的金屬離子中具有最小的離子半徑,而鋰離子篩活性材料小的晶格空位因具有阻礙其他離子半徑較大離子的吸附,而表現出對Li+的高選擇性[20]。目前,應用的電極活性材料主要有LiFePO4、 LiMn2O4和LiNi1/3Co1/3Mn1/3O2等。
2.1 LiFePO4電極
LiFePO4的晶體結構屬于典型的橄欖石型結構(圖1)[21],由FeO6八面體和PO4四面體構成晶體骨架,Li+具有一維的可移動性。Li+在鐵的氧化與還原過程中實現Li+的脫出與嵌入過程。
LiFePO4在實現Li+脫出與嵌入過程中的反應過程分別如式(1)和式(2)所示。
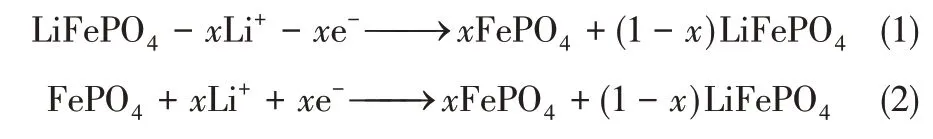
橄欖石型的LiFePO4結構中[LiO6]八面體和[FeO6]八面體之間的[PO4]四面體限制了LiFePO4的體積變化,使得其具有良好的循環穩定性,但同時也限制了Li+在充放電過程中的嵌入和脫出,使得Li+的擴散速率較低。此外,由于其結構中沒有連續的[FeO6]共棱八面體網絡,電子傳導只能通過Fe-O-Fe進行,使其電子電導率也較低,因此在電池的充放電過程中其倍率性能較差,對應其提鋰效率也相對較低。
2.2 LiMn2O4電極
LiMn2O4屬于典型的尖晶石結構,如圖2 所示。圖2(b)和(c)分別為LiMn2O4和Li+脫出后λ-MnO2的三維框架結構,MnO2構成了三維的鋰離子嵌/脫通道,使其相較于LiFePO4具有更高的離子電導率和電子電導率[22],從而具備更好的倍率性能。
LiMn2O4在實現Li+脫出與嵌入過程中的反應過程分別如式(3)和式(4)所示。
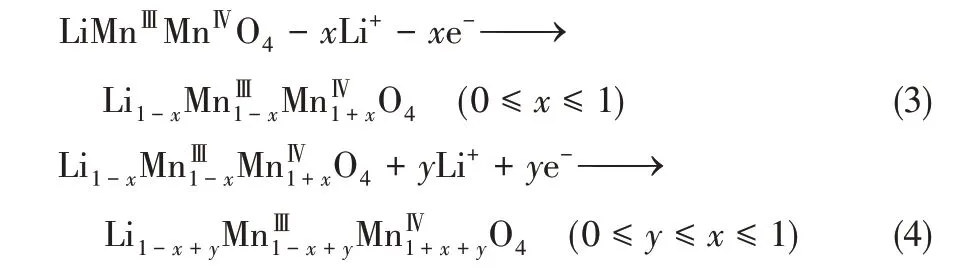
LiMn2O4材料中Mn易溶損,導致其循環穩定性較差,研究者們通過改進其制備方法來提高其循環穩定性[23]。其制備方法主要為高溫固相法和水熱合成法[24]。高溫固相法合成工藝較簡單,適合大批量生產;水熱合成法所得產品純度較高,但操作較為復雜,批量生產受到規模限制。Xu 等[23]用高溫固相法合成LiMn2O4粉末,將其與炭黑、PVDF 按照質量比8∶1∶1 進行混合后涂覆在鉑片上制成LiMn2O4粉末電極,并用水熱合成法制備了自支撐LiMn2O4膜電極。其制備路線是先在陰極電沉積制備Mn(OH)2,通過氧化得到Mn2O3,再在0.025mol/L LiOH 溶液中進行水熱反應得到自支撐的LiMn2O4膜電極。對比兩者的電化學提鋰性能發現,由于高溫固相法得到的LiMn2O4晶體中雜質成分較高,影響了電極的可逆反應,100 次循環后LiMn2O4粉末電極的Li+吸附量為理論值的60.5%,而自支撐LiMn2O4膜電極的Li+吸附量為理論值的82.6%。自支撐LiMn2O4膜電極表現出更好的循環穩定性。
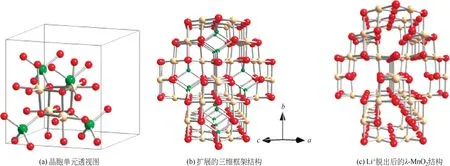
圖2 尖晶石型LiMn2O4結構[22]
2.3 LiNi1/3Co1/3Mn1/3O2 電極
LiNi1/3Co1/3Mn1/3O2為層狀結構,因其高的理論容量、高的充放電倍率和較好的循環穩定性,被認為是理想的正極材料,并被廣泛應用[25]。此外,其在水溶液體系中表現出更快的充放電倍率性能,還具有良好的穩定性,1000 次循環后其容量損失僅有9.1%[26]。
Lawagon 等[27]將其應用于鹵水體系中進行電化學提鋰研究。層狀LiNi1/3Co1/3Mn1/3O2電極的鋰提取與釋放過程示意如圖3所示。在第一步的放電過程中Li+嵌入到電極中,而在第二步的充電過程中,Li+再次釋放到溶液中。在復雜的鹵水體系中,LiNi1/3Co1/3Mn1/3O2電極表現出對Li+的高選擇性。最終獲得的溶液中LiCl純度達96.4%,且過程能耗僅為2.60W·h/mol Li+。
2.4 電極材料對比
基于已有主要研究,對于上述三種電極材料的提鋰性能對比如表1所示。
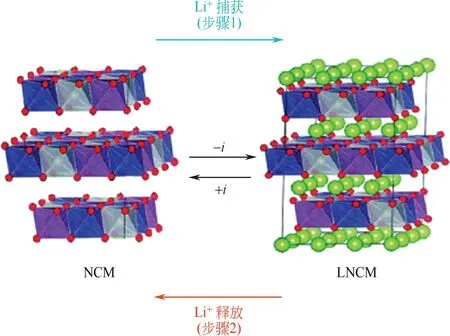
圖3 LiNi1/3Co1/3Mn1/3O2電極提鋰過程[27]
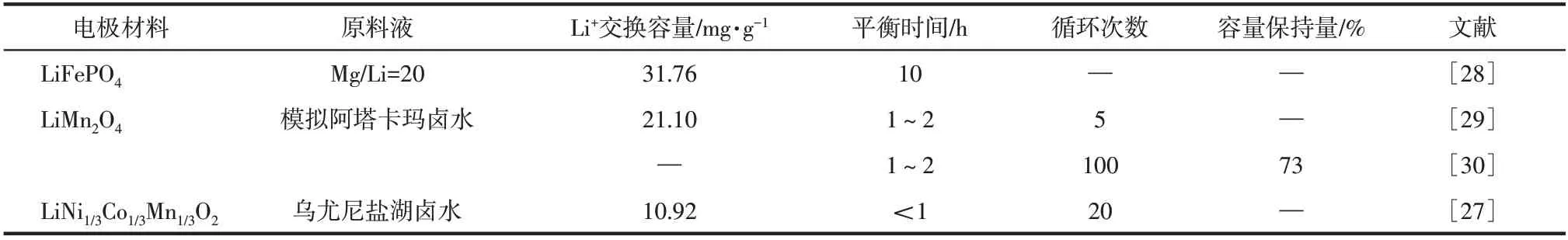
表1 電極材料提鋰性能對比
橄欖石結構的LiFePO4循環穩定性較好,但其電導率較低,提鋰過程中所需的平衡時間較長,效率較低。LiMn2O4制備較為簡單且價格較低,但其在離子嵌脫過程中出現的晶格結構變化使其循環穩定性較差。LiNi1/3Co1/3Mn1/3O2具有較快的離子嵌脫速率和循環穩定性,但其制備條件要求較為苛刻,成本較高。針對目前各電極材料存在的優缺點,通過電極摻雜、包覆等方法對電極材料進行改性,進一步開發出兼具良好選擇性、吸附容量、吸附效率及循環穩定性的新型電極材料,將對電化學提鋰技術的發展及應用起到巨大的推動作用。
3 電化學提鋰體系
除了電極材料外,電化學提鋰體系的構建對電化學提鋰的設備成本、提鋰效率和能耗都有著關鍵的影響。因此,研究者們嘗試通過改進電極體系來促進電化學提鋰的發展。
3.1 金屬對電極體系
3.1.1 Pt對電極體系
Kanoh 等[31]于1993年首次提出利用電化學方法從水溶液中提取Li+資源,組成Pt/λ-MnO2體系進行提鋰,λ-MnO2的嵌鋰容量達到11mg/g。
以λ-MnO2為陰極時發生還原(嵌鋰)反應為式(5)。
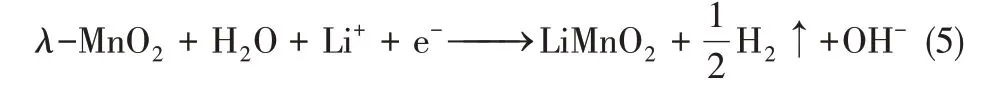
以λ-MnO2為陽極時發生氧化(脫鋰)反應為式(6)。
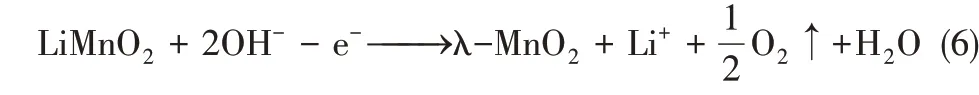
因Pt 電極為惰性電極,故提鋰過程會產生水解副反應,使其能耗偏高,并產生氣體副產物。此外,溶液的pH 條件需進行嚴格控制,以防止共存Mg2+結垢現象的發生。為避免電解水副反應的發生,能捕獲陰離子的對電極被開發,其中Ag/AgCl電極因其與Cl-能發生良好的氧化還原反應而得到廣泛應用。
3.1.2 Ag/AgCl對電極體系
由于Ag/AgCl 電極較為穩定的氧化還原電勢及良好的Cl-結合和釋放性能,避免了電解水副反應的發生,大大降低了過程能耗,提升了電極的循環穩定性,從而使得Ag/AgCl 對電極體系廣泛發展。Pasta 等[32]于2012 年開發了一種由捕獲Li+的電極(LiFePO4)和捕獲Cl-的電極(Ag)組成的LiFePO4/Ag電極體系來進行提鋰過程,主要包括四個步驟,如圖4所示。
第一步,將FePO4電極和Ag 電極放入原料液中,將兩電極短接,FePO4發生還原(嵌鋰)反應,Ag 電極發生氧化(嵌氯)反應,實現氯化鋰的提取,具體反應如式(7)所示。
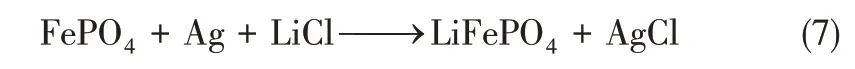
第二步,將反應結束得到的LiFePO4和AgCl放入50mmol/L的KCl回收液中。
第三步,以LiFePO4為陽極、AgCl 為陰極,在兩電極間加入正向直流電場。LiFePO4發生氧化(脫鋰)反應,AgCl發生還原(脫氯)反應,實現LiCl的釋放,具體反應如式(8)所示。
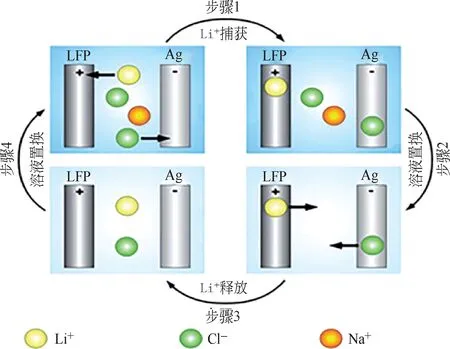
圖4 LiFePO4/Ag電極體系提鋰原理[32]
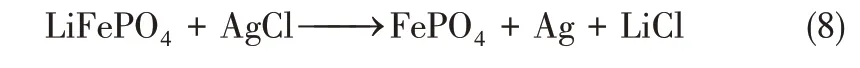
第四步,將反應結束后得到的FePO4和Ag 電極再次放入原料液中,進行第一步反應。
如此重復以上四個步驟,可實現LiCl的選擇性提取。由于步驟一為自發反應過程,因此其不僅不需要消耗能量,還可以釋放能量,其能耗分析如圖5所示。由于步驟一為釋放能量的過程,步驟三為消耗能量過程,因此每次循環過程需要消耗的能量為圖5中陰影部分的面積。
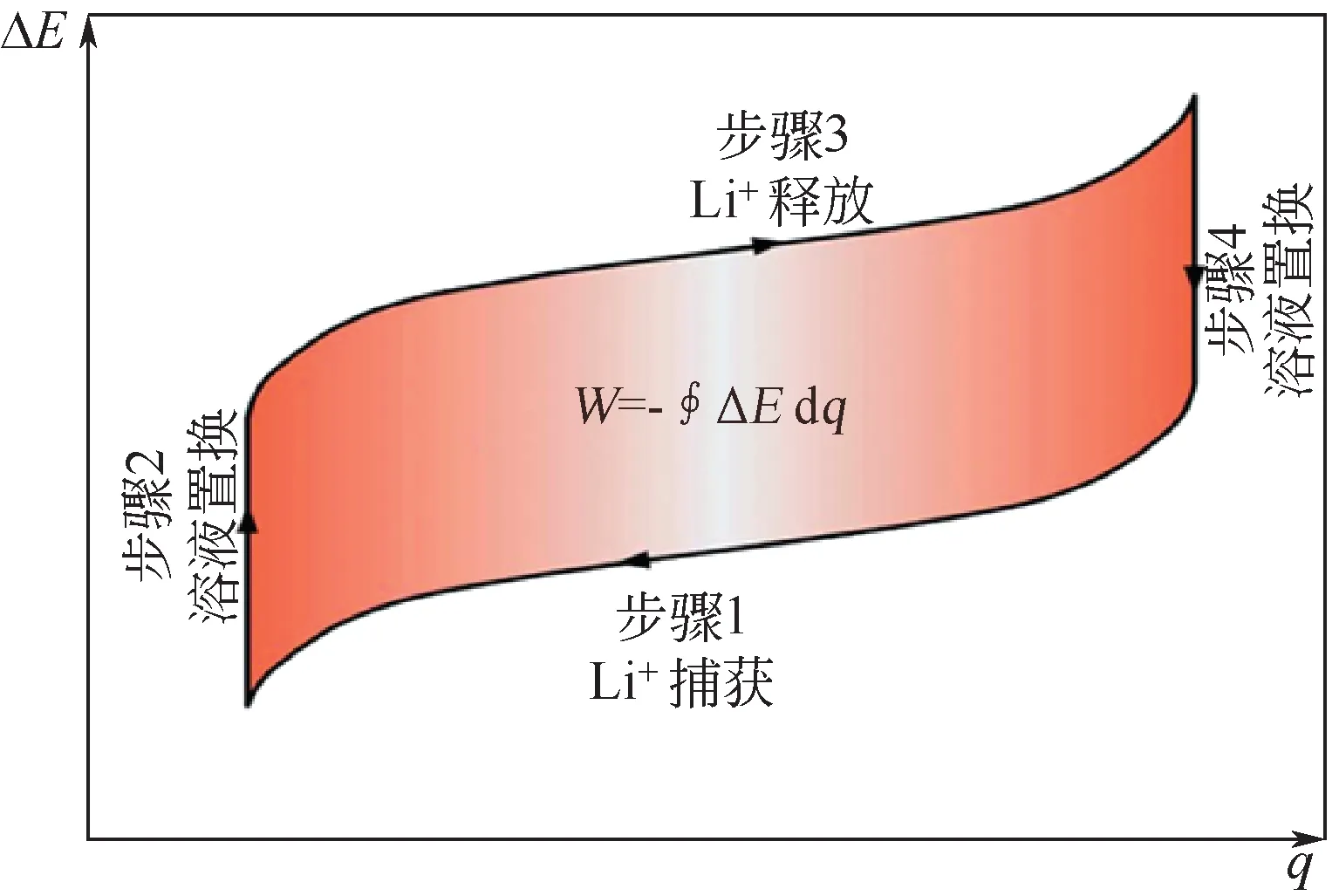
圖5 LiFePO4-Ag電極體系提鋰過程能量消耗[32]
LiFePO4電極對Li+表現出了較高選擇性,LiFePO4/Ag 電極體系可實現將Li∶Na=1∶100 的鹵水中Li+的選擇性提取,得到Li∶Na=5∶1 的溶液,且其能耗僅為144W·h/kg Li。
此外,Jaehan[33]和Lawagon 等[27]分別于2013 年和2018 年開發了λ-MnO2/Ag 和Li1-xNi0.33Co1/3Mn1/3O2/Ag體系用于混合溶液中鋰資源的回收,也達到了避免水電解的低能耗鋰提取反應過程。但貴金屬Ag 電極的使用限制了該電化學提鋰體系的應用及發展。因此,為進一步降低電化學提鋰設備成本,具有經濟性的其他對電極體系逐漸被開發。
3.1.3 Zn對電極體系
Zn 電極相較于Ag 電極而言價格較為低廉,且具有較為穩定的氧化還原電勢。Kim 等[30]構建了LiMn2O4/Zinc 體系來實現LiCl 的選擇性回收,其提鋰過程示意如圖6所示。
如圖6(a)所示,電化學提鋰裝置由陰離子交換膜分隔為兩個室,λ-MnO2電極置于左側原料液中,Zn 電極置于右側ZnCl2溶液中。在鋰提取過程中,將兩電極進行短接,則λ-MnO2進行嵌鋰過程,同時Cl-從原料液中向右遷移至右側,Zn 被氧化為Zn2+。其反應方程如式(9)所示。
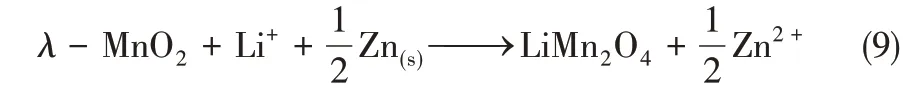
在氯化鋰的回收過程中其反應過程如圖6(b)所示,在LiMn2O4和Zn 電極間施加正向直流電場,LiMn2O4發生氧化反應,Li+從電極上脫出到溶液里;左側溶液中Zn2+發生還原反應,被還原為Zn 單質。其反應方程如式(10)所示。
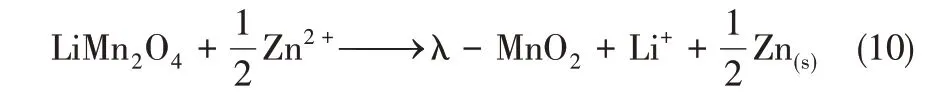
λ-MnO2電極對鋰表現出了較高的選擇性,如圖6(c)所示三次循環后,原料液中Li+濃度的增加量遠大于其他雜質離子;同時,如圖6(d)所示,電極顯示出較強的循環穩定性,在100次循環后,其嵌鋰容量保持初始容量的73%。鋰回收過程能耗為6.3W·h/mol Li。此外,電極對鋰的選擇性系數隨電流密度的增加而降低,在電流密度分別為0.5mA/cm2、0.75mA/cm2和1.0mA/cm2條件下,其選擇性系數分別為14.4、9.2 和6.6;并且隨著電流密度的增加,更多的雜質成分獲得能量嵌入λ-MnO2電極中。
3.2 非金屬對電極體系
3.2.1 活性炭對電極體系
為替代貴金屬甚至金屬對電極,Kim 等[34]用活性炭電極代替了Ag 電極,組成λ-MnO2/活性炭體系(如圖7),降低了設備成本。在活性炭陰極表面負載一張陰離子交換膜以避免充電過程中Li+的嵌入;在陽極和陰離子交換膜中間加一張尼龍隔網組成水通道。
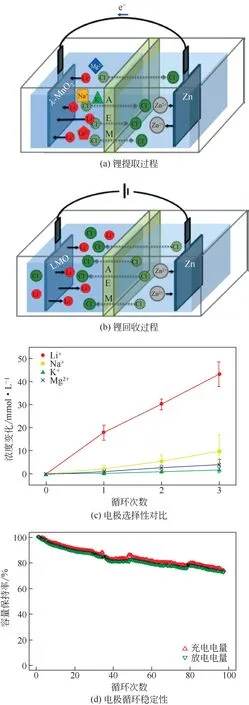
圖6 LiMn2O4/Zinc體系提鋰過程[30]
λ-MnO2/活性炭體系提鋰過程原理如圖8(a)所示。其提鋰過程分為兩步:放電過程即鋰的提取過程,Li+嵌入到λ-MnO2中,Cl-嵌入到活性炭電極中;充電過程即鋰的回收過程,Li+和Cl-分別從LiMn2O4和活性炭電極中脫出,而陰離子交換膜阻擋了Li+嵌入到活性炭電極中,提高了提鋰效率。圖8(b)為充放電過程中離子濃度變化。在放電和充電過程中,Li+濃度分別呈現出了明顯的下降和上升趨勢,而雜質離子的濃度沒有出現明顯變化,從而實現了鋰選擇性提取的目的。此外,該電極體系在循環50 次后,其庫侖效率仍保持在97%以上,電極對Li+選擇性沒有下降,表現出了優異的循環性能。
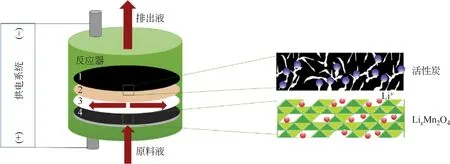
圖7 λ-MnO2/活性炭體系提鋰裝置[34]
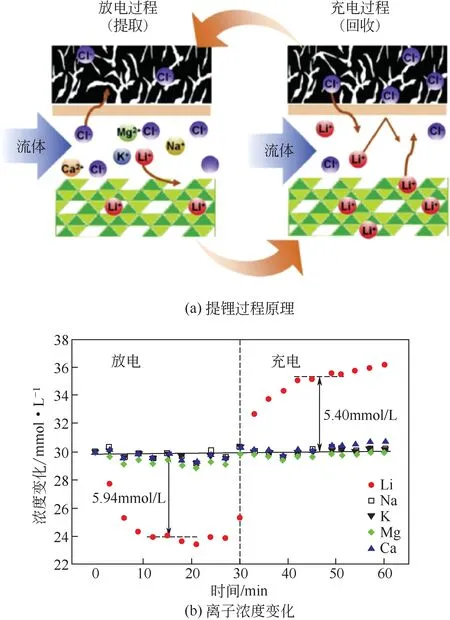
圖8 λ-MnO2/活性炭體系提鋰過程原理和離子濃度變化[34]
3.2.2 聚吡咯對電極體系
聚吡咯(polypyrrole,PPy)是一種高分子導電聚合物,由于其存在共軛π鍵,電子可以在聚合物鏈上進行自由轉移,因此具有良好的導電性,常用來做電極吸附材料或電極材料的改性[35-36]。此外,PPy在氧化還原過程中還具有優異的Cl-可逆交換特性[37]。如圖9 所示,PPy 在還原過程中會釋放Cl-;在氧化過程中會吸附陰離子,并表現出陰離子可逆交換性能。
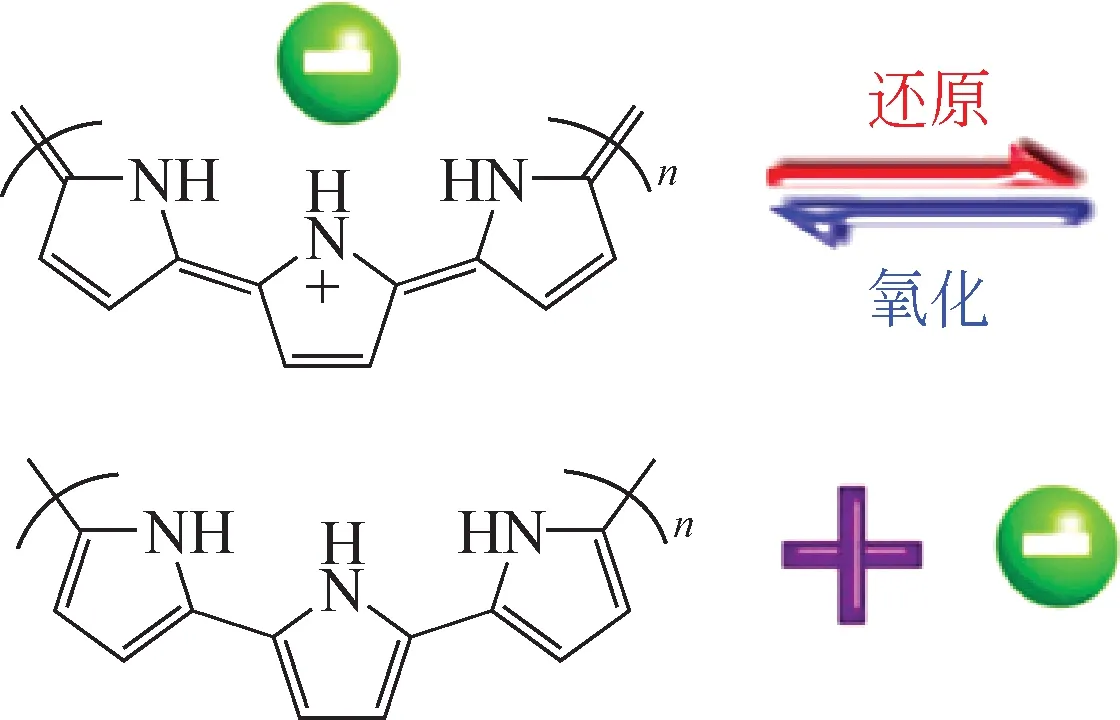
圖9 PPy的陰離子交換機理[37]
基于PPy 的上述特性,Calvo 等[38-40]開發了LiMn2O4/PPy 電極體系來實現LiCl 的回收,其提鋰過程如圖10所示。
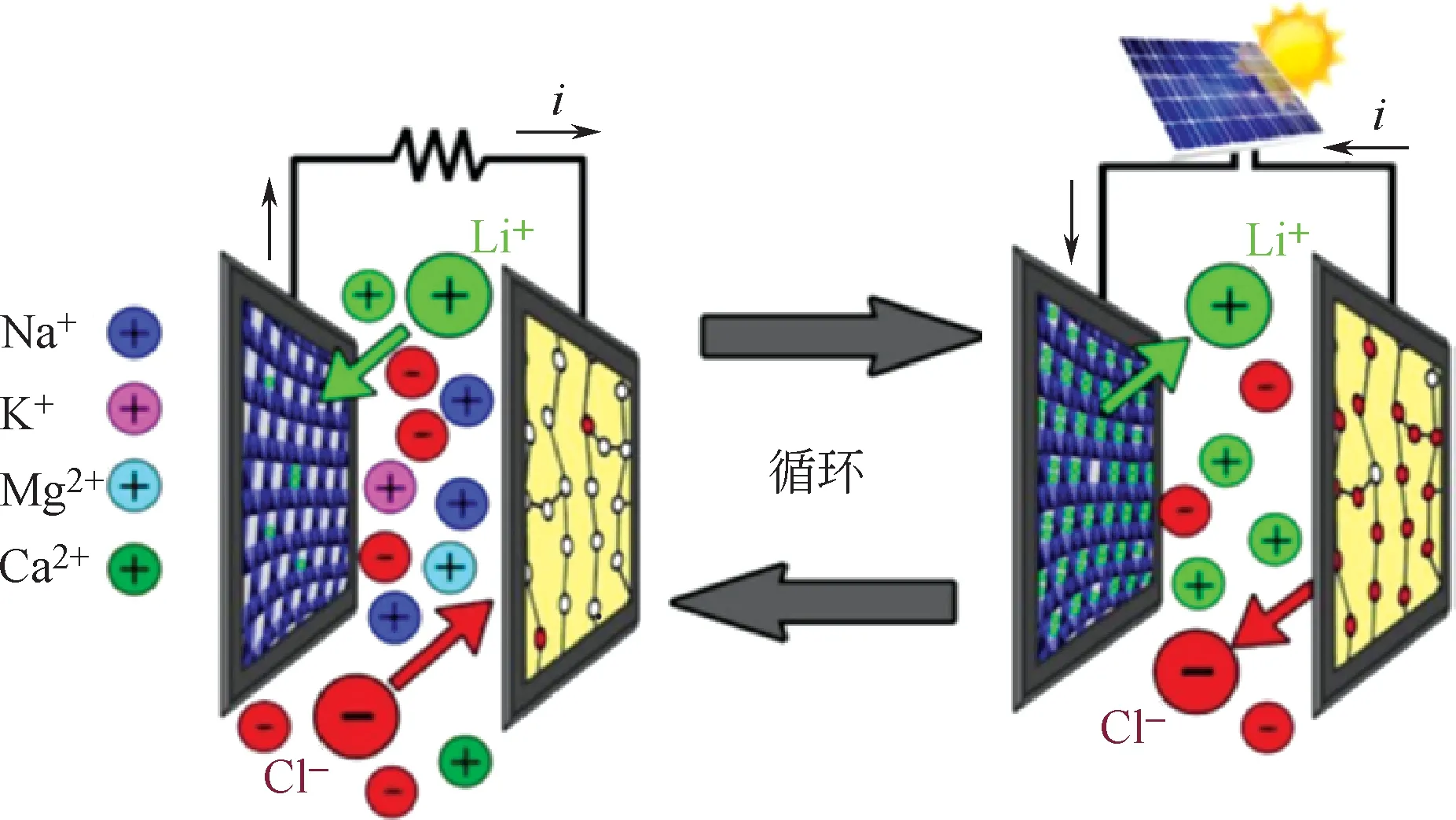
圖10 LiMn2O4/PPy電極體系提鋰過程[40]
兩電極的反應分別如式(11)和式(12)所示。其中,圖10 左側反應為式中的正向反應過程,右側反應為式中的逆向反應過程。

總的反應方程為式(13)所示。
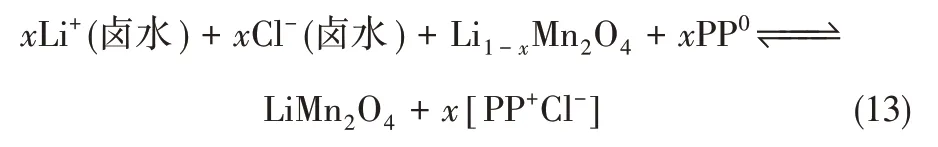
該反應過程的反應電勢低于1V,在200次循環后充電效率保持50%,且其能耗為1mol Li 5~10W·h。
上述電化學提鋰過程均使用了其他材料的對電極,單次只能進行鋰的提取或回收單一過程,提鋰效率較低。為避免對電極的使用,提高提鋰效率,研究者們開發出了“搖椅式”電化學提鋰體系。
3.3 “搖椅式”電極體系
“搖椅式”電極體系是將富鋰態電極和貧鋰態電極組成電極體系。兩電極分別置于由陰離子交換膜分隔的回收室和原料室內,通過施加正向電場,富鋰態電極進行氧化(脫鋰)反應,貧鋰態電極進行還原(嵌鋰)反應,實現脫鋰與嵌鋰過程的同步進行,提高了提鋰效率。
3.3.1 LiFePO4/FePO4電極體系
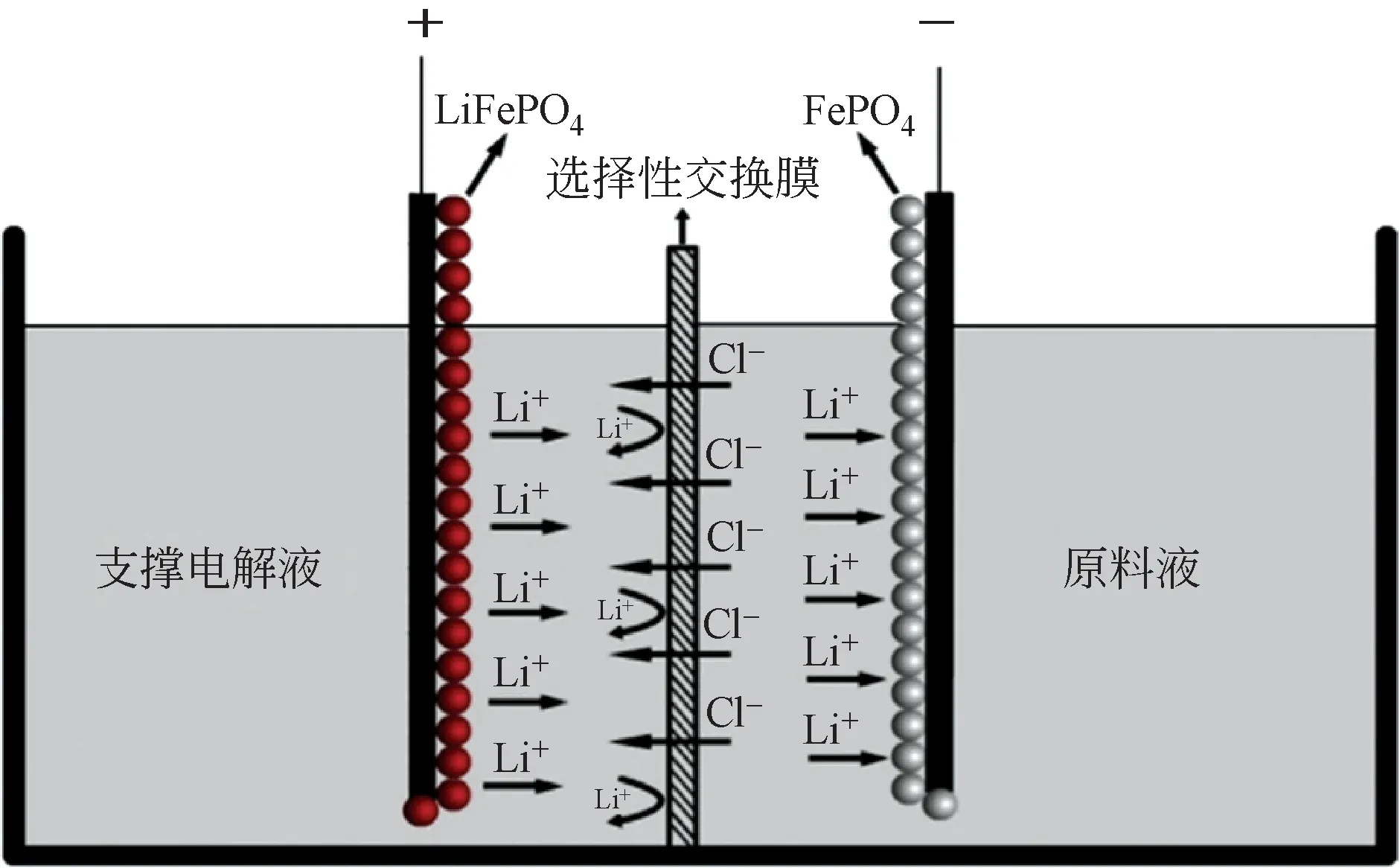
圖11 LiFePO4/FePO4電極體系提鋰原理[42]
中南大學趙中偉教授團隊[41-42]于2013 年開發LiFePO4/FePO4電極體系(如圖11)來實現氯化鋰溶液的選擇性提取。電解槽被陰離子選擇性交換膜分隔為兩個隔室:將LiFePO4電極置于回收溶液(0.5mol/L NaCl)中,FePO4置于原料液(含鋰鹵水)中。在電場下,LiFePO4陽極發生氧化(脫鋰)反應,陰極發生還原(嵌鋰)反應。該過程中原料液中的陰離子(Cl-)會跨過中間的陰離子交換膜遷到回收液中,具體反應如式(14)和式(15)所示。
LiFePO4(陽極)
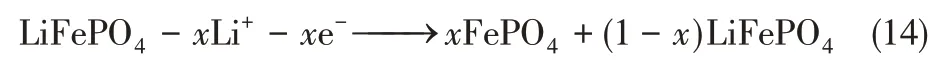
FePO4(陰極)
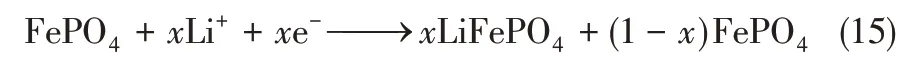
反應結束時LiFePO4電極和FePO4電極分別轉變成FePO4和LiFePO4電極。將兩電極進行位置調換,然后重復上述過程,即可實現氯化鋰的選擇性提取。在純的LiCl溶液中,其嵌鋰量在10h達到最大值38.93mg/g LiFePO4,并將Mg/Li 為60 的鹵水降到了0.45。此外,低的操作電壓更有利于實現鋰與雜質離子間的分離,高的操作電壓則會使更多的雜質離子嵌入到電極中,從而使得電極對Li+的選擇性下降。
3.3.2 LiMn2O4/Li1-xMn2O4電極體系
基于LiMn2O4良好的離子導電性和電子導電性,河北工業大學紀志永教授團隊[29]開發了LiMn2O4/Li1-xMn2O4電極體系(如圖12),并實現了LiCl的選擇性提取。LiMn2O4陽極在直流電場作用下失電子,使得Mn(Ⅲ)被氧化為Mn(Ⅳ),Li+從LiMn2O4電極上釋放到回收液中;Li1-xMn2O4陰極在電場下得電子,使得Mn(Ⅳ)被還原為Mn(Ⅲ),Li+從原料液中嵌入到電極中。當兩電極分別完成其對應的嵌/脫過程后,將兩電極位置進行顛倒,實現鋰資源由原料液側向回收液側的轉移。其陽極和陰極反應分別如式(3)和式(4)所示。此外,Liu 等[43]也圍繞類似體系,就溫度、離子濃度、電壓等條件對提鋰性能影響做了相應探究。
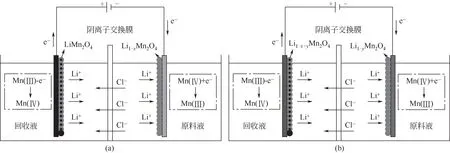
圖12 LiMn2O4/Li1-xMn2O4電極體系提鋰原理[29]
LiMn2O4/Li1-xMn2O4電極體系可以實現鋰資源的高效選擇性提取,在純的LiCl溶液中其提鋰量達到34.31mg/g LiMn2O4。共存離子對提鋰性能的影響順序為Mg2+>Na+>Ca2+>K+,其影響順序由離子半徑大小和離子價態共同決定,小的離子半徑更利于嵌入到電極內部;同時,價態高的離子需要克服更大的水和自由能去水化進入到電極中,從而使得嵌入過程更加困難[44-45]。此外,低的操作電壓更利于實現鋰與共存離子間的分離,與其他研究結果一致[28,30]。在模擬的鹵水提鋰中,其Li+/Na+、Li+/Mg2+和Li+/Ca2+分別上升300倍、70倍和110倍;在模擬濃海水提鋰過程中,其Li+/Na+、Li+/Mg2+和Li+/Ca2+分別上升360 倍、90 倍和105 倍,電流效率達86%。該電極體系可實現鹵水/濃海水中鋰的高選擇性回收,但目前其提鋰能耗(約為20W·h/mol Li)相較于含其他對電極材料還偏高。因此,需要通過提鋰過程及裝備設計優化進一步降低其過程能耗,提高其提鋰性能。
4 結語
電化學提鋰技術是對傳統提鋰方法的發展,在低品位、復雜共存離子的溶液體系下表現出顯著優勢。其中,鋰吸附電極材料及其電化學提鋰體系設計是影響電化學提鋰性能的兩個關鍵因素。對于復雜體系中鋰資源的提取,鋰吸附電極材料的選擇依據主要是吸附容量、選擇性和循環穩定性。目前,LiFePO4、LiMn2O4和LiNi1/3Co1/3Mn1/3O2三種電極材料應用最為廣泛。電化學提鋰體系的設計原則主要為設備成本、提鋰效率和單位能耗等。目前,相比于其他材料對電極體系,“搖椅式”電極體系因其避免對電極的使用,降低了設備成本,提高了提鋰效率而具有一定優勢,但其能耗較含對電極體系還偏高。
電化學提鋰技術目前還存在著一些問題需要被進一步研究解決。針對電極材料的制備,如何在電極制備過程中防止活性物質流失的同時避免過度包埋,如何克服鋰的交換容量較低、電極循環性能較差和高的操作電流/電壓會使得電極對鋰的選擇性下降等不足;針對提鋰體系和裝置設計及提鋰過程,目前“搖椅式”電極體系能耗還偏高,需進一步優化裝置設計及提鋰過程以降低能耗,提高提鋰性能。因此,電化學提鋰技術后續需要在以下幾個方面繼續開展深入系統的工作。
(1)通過調整黏結劑的種類和用量以及功能材料負載量等條件來優化電極制備工藝,在降低乃至防止活性物質流失的同時避免功能材料的過度包埋,以保障乃至進一步提升電化學提鋰效率。
(2)通過電極改性(如摻雜或包覆)制備新型電極材料,提高電極對鋰的交換容量、選擇性和循環性,并克服電極在高的操作電流/電壓條件下選擇性下降的不足。
(3)通過電極材料結構參數的變化,探究電極材料的物性參數對提鋰性能的影響機理,為后續電極材料的設計提供指導。
(4)通過系統探究共存離子及操作條件對提鋰動力學過程的影響,指導優化電化學提鋰過程。
(5)通過對電化學提鋰設備進行優化,提高提鋰過程的自動化水平,提升其提鋰效率,推進電化學提鋰的工業化應用進程。
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
