UPLCMS/MS法同時測定不同動物源性食品中硝基呋喃代謝物殘留
2020-06-17 07:41:10呂珍珍李昌松羅中魏潘志明
食品工業(yè)科技 2020年12期
關(guān)鍵詞:檢測
呂珍珍,程 瀧,李昌松,張 嫻,羅中魏,潘志明,*
(1.德陽市食品藥品安全檢驗檢測中心,四川德陽 618000;2.德陽市食品檢驗重點實驗室,四川德陽 618000;3.西南科技大學(xué)生命科學(xué)與工程學(xué)院,四川綿陽 621010;4.四川華勝農(nóng)業(yè)股份有限公司,四川德陽 618000)
硝基呋喃類藥物是一種被廣泛用于水產(chǎn)業(yè)和畜禽養(yǎng)殖業(yè)的抗菌藥物,對一些革蘭氏陽性菌和革蘭氏陰性菌、某些真菌和原蟲具有抑制和殺滅作用,通常用于預(yù)防和治療禽類因感染沙門氏菌和大腸埃希菌而產(chǎn)生的疾病[1-3]。然而,硝基呋喃類藥物具有潛在的致畸、致癌、致突變作用,其濫用造成動物體內(nèi)獸藥殘留,最終危害食用人群的身體健康[4]。歐盟1995年EC/1442/95將呋喃唑酮列為不得檢出藥物[5-6];我國農(nóng)業(yè)部在2002年發(fā)布1號、193號、235號公告明確禁止呋喃它酮、呋喃唑酮用于所有動物源性食品,規(guī)定其不得檢出,在2005 年的560號公告中將呋喃西林、呋喃妥因列為禁用獸藥[7-8]。
目前,硝基呋喃類藥物的檢測方法有高效液相色譜法[9](High performance liquid chromatography,HPLC)、高效液相色譜串聯(lián)質(zhì)譜法[10-12](Liquid chromatography-tandem mass spectrometry,LC-MS/MS)、酶聯(lián)免疫法[13-14](Enzyme-linked immunosorbent assay,ELISA)、熒光免疫分析法[15](Fluoro immune assay,FIA)等。其中色譜串聯(lián)質(zhì)譜法仍然是主要的檢驗檢測方法。LC-MS/MS具有較高的靈敏度,較好的選擇性和特異性,可以提供待測物的結(jié)構(gòu)信息[16-17]。然而,諸如LC-MS/MS等此類儀器分析方法通常前處理耗時長、成本高[18-19],并且現(xiàn)有研究方法多局限于某一種基質(zhì),因此,非常有必要建立一種準(zhǔn)確快速的不同基質(zhì)中硝基呋喃類藥物殘留的檢測方法。
硝基呋喃類藥物被攝入體內(nèi)后極不穩(wěn)定,其代謝產(chǎn)物包括氨基脲(SEM)、1-氨基乙內(nèi)酰脲(AHD)、3-氨基-2-唑烷基酮(AOZ)、5-甲基嗎啉-3-氨基-2-唑烷基酮(AMOZ)[20-22]。本研究采用超高效液相色譜串聯(lián)質(zhì)譜法(UPLC-MS/MS),以豬肉、豬肝、螃蟹和蝦4種不同的動物源性樣品作為研究對象,優(yōu)化前處理步驟,構(gòu)建用于同時測定硝基呋喃類藥物4種代謝產(chǎn)物的檢測方法,以期為硝基呋喃類藥物等獸藥殘留檢測技術(shù)提供理論支撐和參考。
1 材料與方法
1.1 材料與儀器
新鮮淡水蝦5批、淡水蟹3批、海水蝦4批、海水蟹4批、小龍蝦2批、豬肉42批、豬肝20批 當(dāng)?shù)剞r(nóng)貿(mào)市場;雞肉中呋喃唑酮代謝物(AOZ,特性值7.60 μg/kg、量值范圍3.84~11.35 μg/kg)、呋喃它酮代謝物(AMOZ,特性值7.75 μg/kg、量值范圍2.94~12.56 μg/kg)定量分析質(zhì)控樣品 中國檢驗檢疫科學(xué)研究院測試評價中心;3-氨基-2-惡唑酮(AOZ)、5-嗎啉甲基-3-氨基-2-惡唑烷基酮(AMOZ)、1-氨基-乙內(nèi)酰脲(AHD)、氨基脲(SEM) 標(biāo)準(zhǔn)值100 mg/L,內(nèi)標(biāo)標(biāo)準(zhǔn)儲備液:AOZ-D4、AMOZ-D5、AHD-13C3、SEM-13C15N2,標(biāo)準(zhǔn)值50 mg/L,農(nóng)業(yè)部環(huán)境保護(hù)科研監(jiān)測所;甲醇、乙腈、正己烷、乙酸乙酯 色譜純,德國Merck公司;甲酸(含量85.0%) 韓國德山藥品工業(yè)(株);2-硝基苯甲醛(純度99%) 上海譜振生物科技有限公司;乙酸銨(純度98.0%) 北京百靈威科技有限公司。
液相色譜-串聯(lián)質(zhì)譜儀、Agilent1290超高效液相色譜 美國安捷倫公司;Q-TRAP 4500三重四極桿/復(fù)合線性離子阱質(zhì)譜,配有電噴霧離子源(ESI)、MultiQuantTM定量軟件 美國AB SCIEX公司;UV-R超純水機 法國Millipore公司;TGL-16A臺式高速冷凍離心機 長沙平凡儀器儀表有限公司;VG3 S025渦旋振蕩器、T25高速組織勻漿機 德國IKA公司;MS205DU電子天平、S220pH測定儀 瑞士Mettler-Toledo公司;ZWYR-240恒溫培養(yǎng)振蕩器 上海智城分析儀器制造有限公司;MTN-5800A氮吹濃縮裝置 天津奧特賽恩斯儀器有限公司;VP6122050抽濾裝置 美國Cole-Parmer公司。
1.2 實驗方法
1.2.1 標(biāo)準(zhǔn)溶液配制 硝基呋喃代謝物標(biāo)準(zhǔn)工作液:硝基呋喃代謝物標(biāo)準(zhǔn)儲備液用甲醇稀釋,配制成各組分濃度均為 0.1 μg/mL 的硝基呋喃代謝物標(biāo)準(zhǔn)工作液;硝基呋喃代謝物內(nèi)標(biāo)標(biāo)準(zhǔn)工作液:硝基呋喃代謝物內(nèi)標(biāo)標(biāo)準(zhǔn)儲備液用甲醇稀釋,配制成各組分濃度均為0.1 μg/mL的硝基呋喃代謝物內(nèi)標(biāo)標(biāo)準(zhǔn)工作液;衍生化試劑(0.1 mol/L):準(zhǔn)確稱取1.50 g 2-硝基苯甲醛,用甲醇溶解并定容至100 mL。
1.2.2 樣品水解和衍生化 雞肉中AOZ和AMOZ質(zhì)控樣品開啟真空包裝袋后稱取0.50 g雞肉凍干粉樣品,加入總計1.50 g的蒸餾水?dāng)嚢?5 min混勻,混勻后的樣品為肉糜狀,即為復(fù)原樣品;稱取0.50 g雞肉凍干粉樣品經(jīng)過復(fù)原后等同于2.00 g雞肉糜樣品。豬肉、豬肝、螃蟹、蝦樣品去皮去骨去殼,取可食用部分用組織搗碎機充分打碎混勻,裝入聚乙烯瓶中,于-18 ℃冷凍保存,測定前解凍、稱樣;稱取約2.00 g(精確至0.01 g)的豬肉、豬肝、螃蟹和蝦樣品和復(fù)原后的雞肉糜樣品于50 mL的塑料離心管中,加入8 mL 0.2 mol/L的鹽酸,渦旋震蕩1 min 后,按照最終定容濃度10 ng/mL加入混合內(nèi)標(biāo)標(biāo)準(zhǔn)溶液,加入100 μL 2-硝基苯甲醛溶液,渦旋混合30 s,置37 ℃恒溫培養(yǎng)振蕩器震蕩16 h。
1.2.3 提取和凈化 取出樣品,冷卻至室溫,加入1 mL 0.3 mol/L磷酸鉀溶液,用2 mol/L的氫氧化鈉溶液調(diào)節(jié)pH為7.5±0.5,加入8 mL乙酸乙酯,震蕩提取5 min后,以10000 r/min離心5 min,收集乙酸乙酯層;殘留物用8 mL乙酸乙酯再提取一次,合并乙酸乙酯層。收集液在40 ℃下氮氣吹干,殘渣用1 mL初始流動相充分溶解,再用1 mL乙腈飽和的正己烷液液萃取,棄去正己烷層去除脂肪,下層過0.2 μm濾膜待測。整個操作過程應(yīng)避光進(jìn)行。研究各因素最佳水平時,均吸取0.1 mL濃度為0.1 μg/mL混合外標(biāo)標(biāo)準(zhǔn)溶液和混合內(nèi)標(biāo)標(biāo)準(zhǔn)溶液到豬肉樣品中,作為加標(biāo)。
1.2.4 色譜條件 色譜柱:Agilent Eclipse Plus C18RRHD(2.1 mm×100 mm,1.8 μm);柱溫:30 ℃;進(jìn)樣體積:10 μL;流動相:A為0.1%甲酸水溶液(含0.0005 mol/L乙酸銨),B為乙腈;流速:0.4 mL/min。梯度洗脫條件見表1。

表1 梯度洗脫條件Table 1 Gradient elution conditions
1.2.5 質(zhì)譜條件 電噴霧離子源(ESI),正離子模式掃描,多反應(yīng)監(jiān)測(Multiple reaction monitoring,MRM),電噴霧電壓(IS):5500.0 V,碰撞氣(CAD):Medium,離子源溫度(TEM):550 ℃。4種硝基呋喃類類代謝物及其同位素內(nèi)標(biāo)的多反應(yīng)監(jiān)測離子對見表2。

表2 4種硝基呋喃代謝物衍生物和相應(yīng)內(nèi)標(biāo)物的質(zhì)譜參數(shù)Table 2 MS parameters of four nitrofuran metabolites and corresponding internal standards
1.3 數(shù)據(jù)處理
采用Excel和Origin 8.5進(jìn)行數(shù)據(jù)處理。
2 結(jié)果與分析
2.1 色譜條件的優(yōu)化
本試驗首先對流動相和色譜柱進(jìn)行了優(yōu)化研究。以甲醇和乙腈作為流動相,分別考察在Agilent Eclipse Plus C18RRHD(2.1 mm×100 mm,1.8 μm)和Agilent Eclipse SB C18RRHD(2.1 mm×50 mm,1.8 μm)兩種長度的C18色譜柱中硝基呋喃代謝物衍生物的分離效果和峰形。使用100 mm的C18色譜柱,以乙腈為流動相能獲得更尖銳對稱的色譜峰,且4種代謝物的分離效果更好。正離子模式下,在水溶液中添加一定濃度的甲酸能夠有效地提高分析物的離子化效率,試驗以0.1%的甲酸水溶液(添加0.0005 mol/L乙酸銨)和乙腈對4 種硝基呋喃類代謝物衍生物梯度洗脫,乙腈的初始比例為10%,然后在6 min內(nèi)線性增加至75%,使各化合物在C18柱中保留增強,分離度和峰形良好,靈敏度高,4種硝基呋喃類代謝物衍生物的總離子流圖見圖1;由于動物源性樣品基質(zhì)復(fù)雜,在6.5~9.5 min將乙腈比例增加至95%,防止未知物在色譜柱中蓄積,有利于洗脫出更多雜質(zhì);9.5~13.0 min時間內(nèi),將乙腈的比例從95%降低為初始比例10%,充分平衡色譜柱,為下一針進(jìn)樣做準(zhǔn)備。優(yōu)化后的方法單個樣品進(jìn)樣時間為13.0 min,相比于GB/T 21311-2007、GB/T 20752-2006和農(nóng)業(yè)部781號公告-4-2006三種方法,更加節(jié)約時間,且有效的保護(hù)了色譜柱,為大批量進(jìn)樣提供保障。
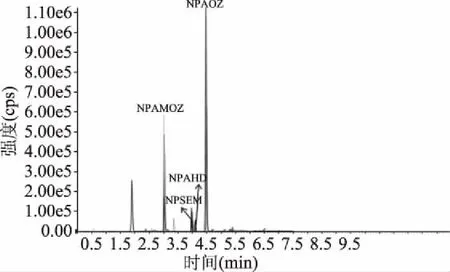
圖1 豬肉樣品中4種硝基呋喃代謝物衍生物總離子流圖Fig.1 Total ion chromatograms of the 4 kinds ofnitrofuran metabolites in porcine muscle
2.2 質(zhì)譜條件的優(yōu)化
在空白豬肉基質(zhì)中添加混合內(nèi)標(biāo)、外標(biāo)標(biāo)準(zhǔn)溶液,按照試驗方法進(jìn)行前處理后上機。選擇ESI+的電離模式,對4種硝基呋喃代謝物及內(nèi)標(biāo)進(jìn)行一級質(zhì)譜掃描,獲得較高響應(yīng)母離子[M+H]+。對去簇電壓和碰撞能量分別進(jìn)行優(yōu)化,使選定的特征碎片離子強度達(dá)到最大。選取2個豐度強且穩(wěn)定的碎片離子作為定性與定量離子,以滿足歐盟657/2002/EC號指令對于禁用藥物液相色譜質(zhì)譜方法定性監(jiān)測的規(guī)定,滿足1個母離子、至少2個子離子共4個識別點數(shù)的要求(見表2)。樣品基質(zhì)中,AHD的m/z為 133.9和103.9兩個碎片離子的響應(yīng)信號較強,很多研究以這兩個碎片離子和準(zhǔn)分子離子峰構(gòu)成監(jiān)測離子對;但是在提取碎片離子色譜圖時發(fā)現(xiàn)豬肉基質(zhì)AHD出峰位置的保留時間±0.3 min內(nèi)左右各有一個小色譜峰。為了進(jìn)一步研究這個現(xiàn)象,分別使用豬肝、蝦、蟹三種基質(zhì)做添加,以及不加樣品基質(zhì)直接水解衍生進(jìn)行上機測試。試驗發(fā)現(xiàn),不加樣品基質(zhì)、蝦、蟹提取AHD的133.9和103.9兩個碎片離子只提取出一個色譜峰,豬肝樣品基質(zhì)中提取AHD的133.9和103.9兩個碎片離子仍然有三個色譜峰,和豬肉樣品一樣,對豐度強且穩(wěn)定的碎片離子再次進(jìn)行提取,發(fā)現(xiàn)m/z為178.0的碎片離子只提取出一個AHD色譜峰,因此,以m/z為133.9、103.9和178.0這三個碎片離子和準(zhǔn)分子離子249.0構(gòu)成監(jiān)測離子對,確保得到準(zhǔn)確的定性定量結(jié)果。
2.3 樣品洗滌液的選擇
GB/T 21311-2007、GB/T 20752-2006和農(nóng)業(yè)部781號公告-4-2006三種方法中都采用甲醇水對樣品進(jìn)行洗滌,以除去部分蛋白質(zhì)、脂肪等雜質(zhì),在相關(guān)文獻(xiàn)中尚未發(fā)現(xiàn)對該部分的研究。本試驗加標(biāo)后依據(jù)樣品處理過程進(jìn)行試驗。比較了不使用洗滌液和不同比例的水與甲醇洗滌液對回收率的影響。水與甲醇的比例分別設(shè)置為1∶1、1∶2、1∶4、1∶8和1∶10,結(jié)果如表3所示,不同比例的水與甲醇洗滌液和不使用洗滌液,加標(biāo)回收均在89.5%~95.8%之間,且差別不明顯。因此,試驗最終選擇不使用洗滌液,能夠有效的節(jié)約時間和成本。
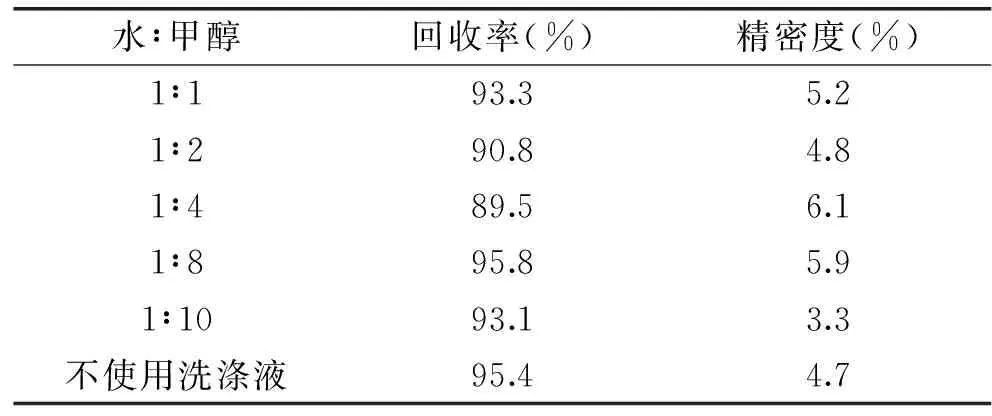
表3 不同比例的水和甲醇洗滌液對回收率的影響(n=6)Table 3 Effects of different proportions ofwater and methanol on the recovery(n=6)
2.4 水解衍生化后pH的調(diào)節(jié)
內(nèi)源性硝基呋喃代謝物以蛋白結(jié)合物的形態(tài)存在于樣品中,經(jīng)鹽酸水解,釋放出硝基呋喃代謝物,同時加入2-硝基苯甲醛衍生16 h。在酸性條件下,硝基呋喃代謝物的含氮親核基團(tuán)胺基(-NH2)與衍生劑的酮醛基團(tuán)(-CHO)基團(tuán)發(fā)生醛胺親和加成反應(yīng)[23]。衍生化后,4種代謝物衍生物的分子離子質(zhì)荷比增大至m/z 200以上,適合質(zhì)譜檢測。在水解衍生化后進(jìn)行萃取操作時,溶液的pH對回收率影響很大。本研究設(shè)計了pH分別為5.0、5.5、6.0、6.5、7.0、7.5、8.0、8.5的8個梯度進(jìn)行試驗,其余試驗操作同1.2。結(jié)果如圖2,pH在7~8的范圍內(nèi)回收率最高,pH偏低或過高均會影響回收率。pH偏低產(chǎn)生的影響更大,這可能是由于pH過低,在酸性條件下衍生物含-N、-NH2形成鹽酸鹽,無法萃取出來。因此,在水解衍生化后pH應(yīng)調(diào)節(jié)在7.5±0.5的范圍內(nèi),最有利于得到更好的回收。
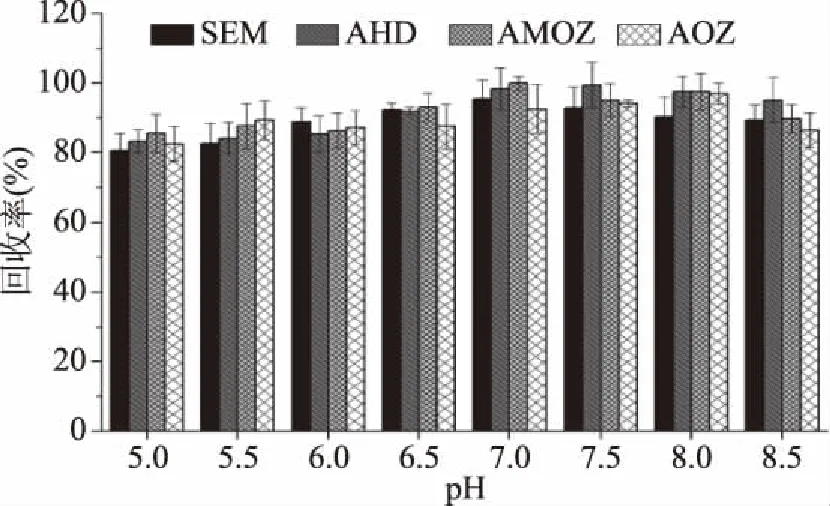
圖2 水解衍生后不同pH得到的4種硝基呋喃代謝物回收率Fig.2 Recoveries of 4 kinds of nitrofuranmetabolites at different pH values
直接用2 mol/L的氫氧化鈉溶液調(diào)節(jié)pH至7.5±0.5較花費時間,先加入1 mL(0.3 mol/L)磷酸鉀形成緩沖體系,再用2 mol/L的氫氧化鈉溶液調(diào)節(jié)pH更加容易。在調(diào)節(jié)pH時,一定要充分渦旋水解衍生化后的混合溶液,并且要用玻璃棒擠壓樣品基質(zhì),使其中的酸被充分釋放出來。通過上述過程對溶液pH進(jìn)行調(diào)節(jié),能夠保證獲得更好的基質(zhì)標(biāo)線和更高的回收率。
2.5 提取次數(shù)對硝基呋喃類代謝物回收率的影響
采用更少提取次數(shù)得到更高的回收率能夠有效的節(jié)約試劑和時間。本研究加標(biāo)后依據(jù)樣品處理過程進(jìn)行試驗。比較了分別用8 mL乙酸乙酯進(jìn)行1次、2次、3次提取(n=6),發(fā)現(xiàn)用8 mL乙酸乙酯進(jìn)行1次、2次、3次提取得到的平均回收率分別為85.1%、96.1%和96.5%,精密度分別為6.5%、5.8%和4.9%。其中乙酸乙酯提取2次得到的平均回收率最高,這可能是由于1次提取并沒有將所有的硝基呋喃類代謝物衍生物提取出來;3次提取與2次提取回收率差別并不大,因此,試驗最終選擇用8 mL乙酸乙酯進(jìn)行2次提取即可得到最佳的回收率。
2.6 定容溶液對檢測的影響
定容溶液可能產(chǎn)生溶劑化作用,影響某些化合物在色譜柱中的保留效果、離子化效率等,進(jìn)而影響峰形和靈敏度。試驗比較了0.1%甲酸水溶液、0.1%甲酸水溶液(含0.0005 mol/L乙酸銨)和梯度洗脫表中初始比例的流動相3種溶液定容。發(fā)現(xiàn)0.1%甲酸水溶液作為定溶液時,SEM的響應(yīng)低;AHD峰分叉,與左右兩個小峰無法分開。0.1%甲酸水溶液(含0.0005 mol/L乙酸銨)作為定溶液時,SEM的響應(yīng)較甲酸水溶液作為定溶液時略有提高,但AHD峰情況相同。采用梯度洗脫表中初始比例的流動相定容,SEM的響應(yīng)最高,且AHD峰峰形較好,能夠與其它雜峰有效分離,結(jié)果見圖1。這與趙東豪等[24]研究結(jié)果一致。
2.7 標(biāo)準(zhǔn)工作曲線、檢出限和定量限
本試驗分別采用4種硝基呋喃代謝物的同位素標(biāo)記物作為內(nèi)標(biāo)物,能夠有效減少各操作步驟對回收率的影響,并且采用試驗驗證為未檢出的陰性樣品基質(zhì)配制標(biāo)準(zhǔn)曲線,使定量更加準(zhǔn)確。稱取5份約2.00 g(精確至0.01 g)的陰性樣品于50 mL的塑料離心管中,加入8 mL 0.2 mol/L的鹽酸,渦旋震蕩1 min后,按照最終定容濃度1、5、10、50、100 ng/mL加入混合標(biāo)準(zhǔn)工作液,按照最終定容濃度10 ng/mL加入混合內(nèi)標(biāo)標(biāo)準(zhǔn)工作液,余下操作同1.2實驗方法。以各定量離子離子質(zhì)量色譜峰面積與相應(yīng)內(nèi)標(biāo)物的峰面積比為縱坐標(biāo),對照溶液的濃度為橫坐標(biāo)制作標(biāo)準(zhǔn)曲線,各回歸方程和相關(guān)系數(shù)如表4所示。回歸方程及相關(guān)系數(shù)表明,采用本方法對4種不同的樣品基質(zhì)中硝基呋喃代謝物進(jìn)行檢測,在 1~100 ng/mL范圍內(nèi)線性關(guān)系良好。以3倍信噪比(S/N)計算得到檢出限(LOD),10倍信噪比(S/N)計算定量限(LOQ)得到在各種樣品基質(zhì)中,AOZ的檢出限為0.1 μg/kg、定量限為0.4 μg/kg,SEM和AMOZ的檢出限為0.2 μg/kg、定量限為0.5 μg/kg,AHD的檢出限均為0.5 μg/kg、定量限為1.0 μg/kg。
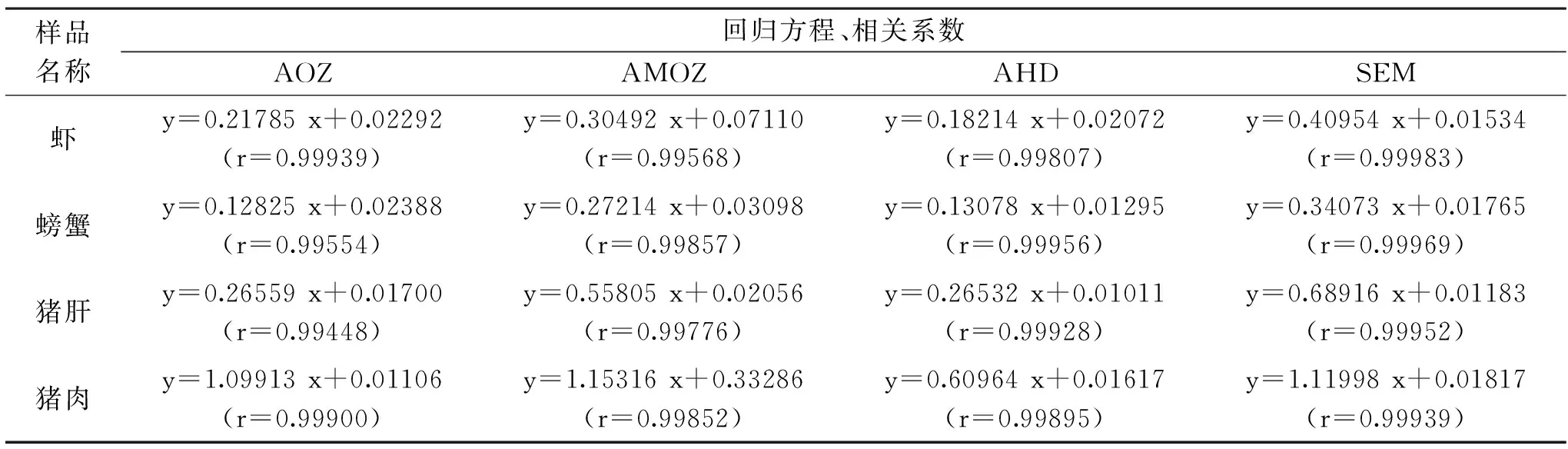
表4 4種樣品基質(zhì)中4種硝基呋喃代謝物的回歸方程Table 4 Regression equations of four nitrofuran metabolites in four samples
2.8 方法回收率和精密度
在4種陰性樣品基質(zhì)中分別加入硝基呋喃代謝物混合標(biāo)準(zhǔn)溶液,使樣品中終含量為1、2和5 μg/kg,按照最終定容濃度10 ng/mL加入混合內(nèi)標(biāo)標(biāo)準(zhǔn)溶液。參照樣品處理過程進(jìn)行標(biāo)回收率試驗,每個添加水平做6次平行樣品處理及上機測定,回收率結(jié)果和相對標(biāo)準(zhǔn)偏差RSD結(jié)果見表5。由表5可以看出,4種代謝物的平均回收率在84.6%~118.1%之間;相對標(biāo)準(zhǔn)偏差RSD在2.9%~8.1%之間。根據(jù)GB/T 27404-2008《實驗室質(zhì)量控制規(guī)范食品理化檢測》中回收率的相關(guān)規(guī)定,被測組分含量小于0.1 mg/kg時,回收率范圍在60%~120%內(nèi)實驗結(jié)果可信。
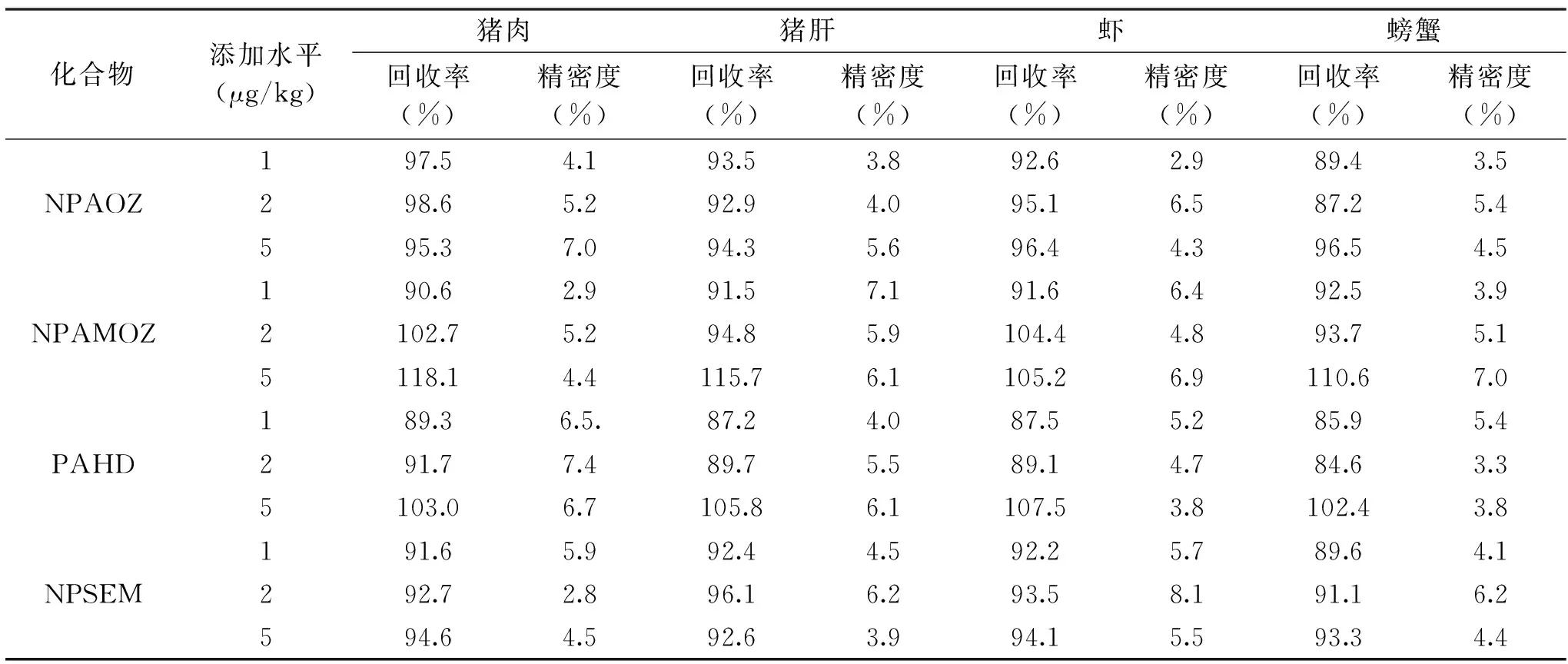
表5 4種樣品基質(zhì)中4種硝基呋喃代謝物的回收率和精密度Table 5 Recoveries and RSDs of four nitrofuran metabolites spiked in four blank samples
2.9 實際樣品的檢測
為了進(jìn)一步驗證方法的可靠性,采用本方法和國家標(biāo)準(zhǔn)方法GB/T 20752-2006分別對市場上隨機購買的80份動物源性樣品和雞肉中硝基呋喃代謝物質(zhì)控樣進(jìn)行檢測。樣品分布包括淡水蝦5批、淡水蟹3批、海水蝦4批、海水蟹4批、小龍蝦2批、豬肉42批、豬肝20批,檢測結(jié)果如表6所示。除表6所列4個樣品檢出陽性,其余樣品均為未檢出,檢出率達(dá)到5%。可以看出,硝基呋喃代謝物檢出的項目主要為AOZ和SEM,且主要集中在淡水蝦、淡水蟹兩個品種。通過國家食品藥品監(jiān)督管理總局網(wǎng)站查詢發(fā)現(xiàn)硝基呋喃類不合格的農(nóng)產(chǎn)品中主要也是AOZ和SEM,集中于水產(chǎn)品。這可能是由于AOZ可用于羅氏沼蝦、鰻鱺、對蝦和河蟹等水生動物養(yǎng)殖環(huán)境的消毒藥物,還可用于防治畜禽腸道感染;SEM一般用作外用消毒藥物,也有拌餌投喂防治水生動物疾病的作用[25]。考慮到硝基呋喃代謝物對人體的影響,對硝基呋喃代謝物進(jìn)行快速準(zhǔn)確的檢測具有非常重要的意義。使用本方法與國標(biāo)方法GB/T 20752-2006測定結(jié)果相近,質(zhì)控樣的檢測結(jié)果都在量值范圍內(nèi),表明本方法能夠滿足硝基呋喃代謝物的定性定量篩查,且本方法不需要使用國家標(biāo)準(zhǔn)中的Oasis HLB固相萃取柱,能夠有效的節(jié)約成本和時間。
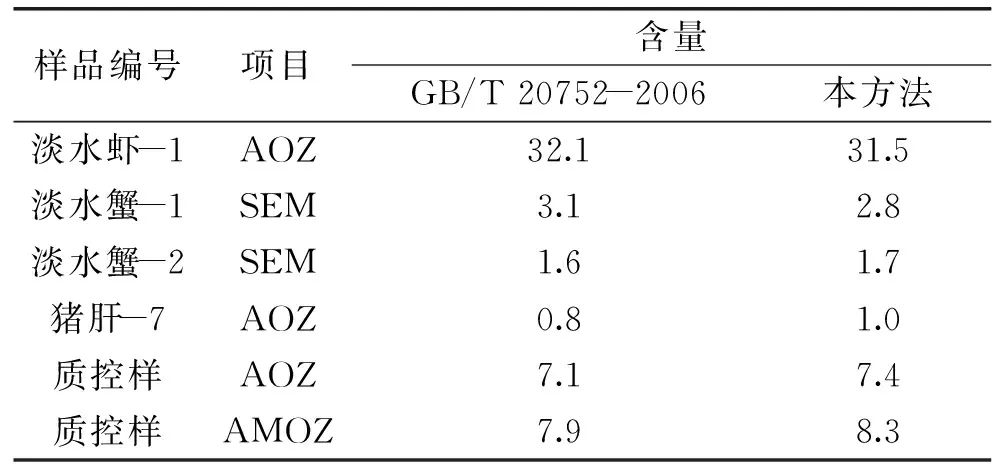
表6 陽性樣品檢測結(jié)果(n=2,μg/kg)Table 6 Detection results of positive samples(n=2,μg/kg)
3 結(jié)論
現(xiàn)有硝基呋喃代謝物檢測的研究主要針對單一動物源性樣品,本研究探索了不同動物源性樣品基質(zhì)中硝基呋喃代謝物的檢測。結(jié)果表明本方法在1~100 ng/mL代謝物濃度范圍內(nèi)線性關(guān)系良好,回收率、精密度、檢出限和定量限均滿足國家標(biāo)準(zhǔn)要求,可用于肉、豬肝、螃蟹和蝦4種動物源性樣品基質(zhì)中硝基呋喃代謝物的檢測。相比于國標(biāo)方法,本方法優(yōu)化了樣品前處理的條件以及UPLC-MS/MS儀器條件,更加節(jié)約時間和成本,適用于大批量檢測不同品種的動物源性樣品。本方法還可進(jìn)一步探究其在其他動物源性樣品基質(zhì)中硝基呋喃代謝物檢測的可行性。
猜你喜歡
中國設(shè)備工程(2022年12期)2022-07-11 04:33:00
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2020年12期)2021-01-18 06:57:46
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2019年9期)2019-11-25 07:34:36
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2019年9期)2019-11-25 07:34:34
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2019年12期)2019-05-21 02:53:50
中學(xué)生數(shù)理化·七年級數(shù)學(xué)人教版(2019年12期)2019-05-21 02:53:48
