水熱法一步合成磷酸鐵鋰及其性能研究
2020-06-12 03:34:50賈雙珠
無機鹽工業(yè) 2020年6期
郭 舉,賈雙珠
(1.茅臺學院,貴州仁懷564507;2.黔南民族師范學院;3.貴州大學;4.中低品位磷礦及其共伴生資源高效利用國家重點實驗室)
Padhi 等[1]于1997 年首次報道了具有可逆脫出、嵌入鋰離子能力的LiFePO4正極材料, 引起了廣泛的關(guān)注和大量的研究。 與鈷酸鋰、錳酸鋰、鎳鈷鋁酸鋰等其他鋰電池正極材料相比,LiFePO4具有原料來源廣泛、價格低廉、比容量高、放電平臺穩(wěn)定、安全性高等優(yōu)點[2],現(xiàn)已成為商業(yè)應(yīng)用最為廣泛的鋰離子電池正極材料之一。 研究發(fā)現(xiàn), 橄欖石結(jié)構(gòu)的LiFePO4晶體由FeO6八面體、LiO6八面體、PO4四面體共同組成, 在其晶胞結(jié)構(gòu)中氧原子近似于立方緊密堆積,磷原子在氧四面體的4c位,鐵原子在氧八面體的4c位,Li+在4a位且平行于c軸[3],這種結(jié)構(gòu)導致了鋰離子較低的擴散系數(shù)和電子傳導速率[4],分別只有1×10-14cm2/s、1×10-9S/cm2[5],影響了LiFePO4材料的電化學性能,可通過表面碳包覆、納米化材料尺寸、摻雜金屬離子等[6]改性手段提高材料的性能。而材料的合成技術(shù)有助于控制好材料顆粒的尺寸、形貌、結(jié)構(gòu)等指標,是提高材料性能的重要改性手段。 因此,對材料合成技術(shù)的研究具有極其重要的研究價值和意義。
目前,LiFePO4的合成方法主要有固相法和液相法。 固相法[7]是將原材料按照一定的比例在一定條件下進行固相反應(yīng)制備LiFePO4, 主要包括高溫固相法、碳熱還原法等。 液相法是相對于固相法而言,是在液相中進行的合成方法,主要包括水熱法、溶膠-凝膠法等[8]。 固相法具有工藝簡單、易于工業(yè)化等特點,但是在材料形貌、粒度控制方面不如液相法[9],這些指標正是材料改性關(guān)注的重點,對材料性能的提升有重要作用,而液相法恰恰能彌補這一缺點。但是,目前的研究[10-11]大多是采用兩步法合成LiFePO4,即先利用液相法合成磷酸鐵前驅(qū)體,然后再利用固相法合成LiFePO4材料, 在固相反應(yīng)過程中材料可能會有晶粒二次生長現(xiàn)象[7],使得材料的結(jié)構(gòu)、形貌、粒度等在一定程度上仍然受固相法的影響,沒有完全發(fā)揮出液相法的傳質(zhì)、傳熱快以及對形貌、粒度等控制的優(yōu)勢,有可能對材料的性能造成影響。
為優(yōu)化液相法一步制備LiFePO4材料技術(shù),筆者研究了液相水熱法一步合成LiFePO4材料及其性能。 以七水合硫酸亞鐵、磷酸二氫銨、一水合氫氧化鋰為原料, 采用液相水熱法合成技術(shù), 通過添加SDBS 作為表面活性劑,抗壞血酸作為抗氧化劑,優(yōu)化液相水熱法合成LiFePO4材料的組成、 結(jié)構(gòu)、形貌、粒度等指標,合成得到了微米級球形顆粒形貌的正交晶系LiFePO4, 并組裝成CR2032 扣式電池,研究了材料的電化學性能。
1 實驗部分
1.1 實驗原料
一水合氫氧化鋰、七水合硫酸亞鐵、磷酸二氫銨、抗壞血酸、十二烷基苯磺酸鈉,均為分析純。
1.2 材料的合成方法
按照物質(zhì)的量比[n(Li)∶n(Fe)∶n(P)=3∶1∶1]稱取一水合氫氧化鋰、七水合硫酸亞鐵、磷酸二氫銨,分別配制成質(zhì)量分數(shù)為10%、15%、25%的水溶液。 在攪拌條件下將3 種水溶液緩慢混合,并添加磷酸鐵鋰產(chǎn)品理論產(chǎn)量2%(質(zhì)量分數(shù))的表面活性劑SDBS和30%(質(zhì)量分數(shù))的抗壞血酸,混合攪拌0.5 h,調(diào)節(jié)pH 為7.0~7.5,得到前驅(qū)體溶液。將前驅(qū)體溶液置于密閉高壓水熱反應(yīng)釜(2 L)中,在180 ℃水熱反應(yīng)10 h。 冷卻后洗滌、抽濾,在真空干燥箱中于105 ℃干燥2 h,冷卻后研磨,得到LiFePO4樣品。
1.3 材料表征
采用Optima 7000 型電感耦合等離子體發(fā)射光譜儀(ICP-OES)對Fe、P、Li 等元素含量進行定量分析;利用X′Pert Powder Ⅲ型X 射線衍射儀(XRD)表征產(chǎn)品的物相結(jié)構(gòu);利用SIGMA-500 型場發(fā)射掃描電鏡(SEM)觀察產(chǎn)品的形貌;利用Mastersizer 3000型激光粒度儀測試產(chǎn)品的粒度。
1.4 電化學性能測試
以合成的LiFePO4材料作為活性物質(zhì), 乙炔黑為導電劑,聚偏氟乙烯為粘接劑,按照質(zhì)量比為75∶15∶10 并用N-甲基-2-吡咯烷酮為溶劑調(diào)成料漿涂于鋁箔上,真空120 ℃干燥12 h,制成圓片作為正極片。以鋰片作為負極,1 mol/L LiPF6的碳酸乙烯酯/碳酸二甲酯(體積比為1∶1)溶液作為電解液,聚丙烯微孔膜作為電池的隔膜,在手套箱內(nèi)組裝成電池。控制手套箱環(huán)境為氬氣氛圍且氧體積分數(shù)低于1×10-5、水分質(zhì)量濃度不超過2.5×10-6mg/L,組裝成CR2032扣式電池。紐扣電池在充滿氬氣的Lab-2000 型手套箱中進行密封裝配。
采用武漢藍電電池測試系統(tǒng)(LAND-BT2013A型)在室溫下進行不同倍率恒流充放電和循環(huán)性能測試, 測試電壓范圍為2.0~4.2 V, 充放電倍率為0.1C、1C、5C、10C。采用上海辰華CHI760D 型電化學工作站測試交流阻抗(EIS),所選頻率為100 kHz~10 mHz,激發(fā)電壓為5 mV。
2 結(jié)果與討論
2.1 產(chǎn)品的組成及結(jié)構(gòu)分析
利用ICP-OES 對合成的LiFePO4材料進行元素定量分析,結(jié)果見表1;利用XRD 對產(chǎn)品的物相結(jié)構(gòu)進行表征,結(jié)果見圖1;對測得的XRD 數(shù)據(jù)進行擬合,計算得到產(chǎn)品的晶胞參數(shù)及晶胞體積,結(jié)果見表2。
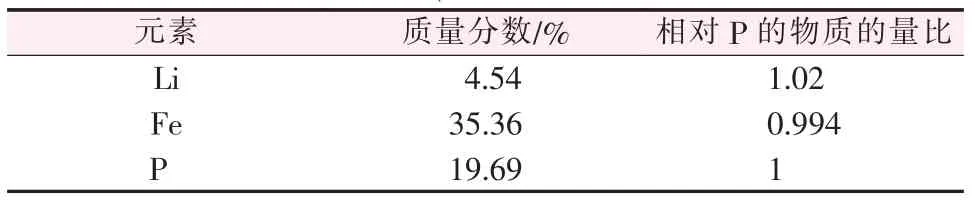
表1 LiFePO4 材料元素含量分析
從表1 可知, 合成的LiFePO4材料中Li、Fe、P元素質(zhì)量分數(shù)分別為4.54%、35.36%、19.69%。 以P元素為參照標準對應(yīng)計算得到Li、Fe、P 元素物質(zhì)的量比為1.02∶0.994∶1,這一數(shù)值并非嚴格符合LiFePO4材料理論物質(zhì)的量比1∶1∶1。 這是由于,在水熱法一步合成LiFePO4材料過程中,添加過量的Li+占據(jù)了部分原Fe2+位,造成一定的晶格錯位[12-13],合成得到了Li1.02Fe0.994PO4材料。 這一推論可從表2 晶胞參數(shù)數(shù)據(jù)顯示的合成材料的晶胞體積輕微膨脹得到證明。 這種非理論化學計量比的LiFePO4材料,由于Li+占據(jù)了部分Fe2+位,使得Li+在晶格間的擴散阻力減小,擴散速率加快,有利于提高材料的電化學性能[14]。
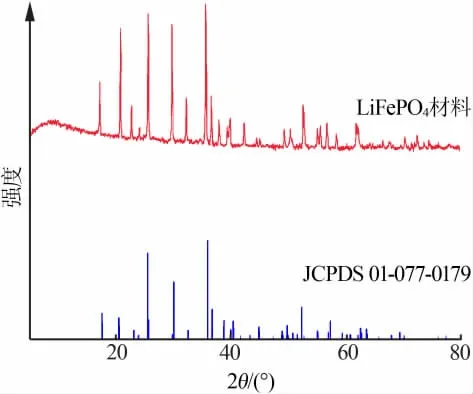
圖1 LiFePO4 材料XRD 譜圖
從圖1 可知, 合成的LiFePO4材料主要特征峰與LiFePO4標準譜圖(JCPDS 卡01-077-0179)基本一致,沒有明顯雜峰,顯示產(chǎn)品為純相的正交晶系橄欖石結(jié)構(gòu)的LiFePO4,空間群為Pnma。 樣品XRD 譜圖也表明,微過量的Li+錯位占據(jù)部分Fe2+位并沒有對材料的結(jié)構(gòu)產(chǎn)生影響,這與Park 等[13]的研究結(jié)論一致。 LiFePO4材料在水熱合成過程中添加了一定量的SDBS 作為表面活性劑,但是從樣品XRD 譜圖上并沒有看到C 的衍射峰,說明表面活性劑在產(chǎn)品洗滌的過程中被完全洗去。
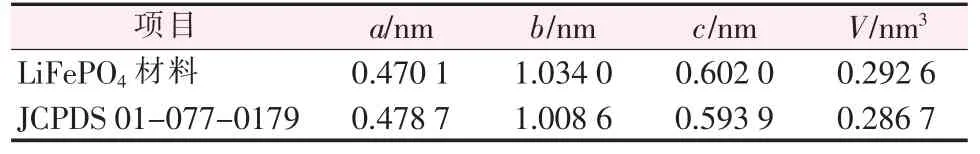
表2 LiFePO4 材料晶胞參數(shù)及晶胞體積
從表2 可知,合成的LiFePO4材料與LiFePO4標準物質(zhì)(JCPDS 卡01-077-0179) 相比晶胞參數(shù)沿b、c軸均有所增加,使得晶胞體積略有增加。 這是由于,Li+有效離子半徑為0.076 nm[9],而Fe2+有效離子半徑為0.074 nm[13],隨著Li+占據(jù)Fe2+位增多,晶胞參數(shù)a、b、c軸數(shù)值逐漸增大,理論上將會造成晶胞體積有一定的膨脹。 此推論與實際檢測到的數(shù)據(jù)相符,合成材料的晶胞體積為0.292 6 nm3,而標準卡對應(yīng)的LiFePO4晶胞體積為0.286 7 nm3。
2.2 產(chǎn)品的形貌和粒度分析
利用SEM 觀察LiFePO4材料的微觀形貌,結(jié)果見圖2;在樣品SEM 照片基礎(chǔ)上,結(jié)合激光粒度儀對樣品粒度進行分析,結(jié)果見圖3。 圖2a、b、c、d 分別為樣品在不同放大倍數(shù)下的SEM 照片。 由圖2 可知,樣品為球形顆粒形貌,且球形度較高,形貌規(guī)整。 球形形貌有利于提高材料的比表面積、 增強電化學反應(yīng)活性、降低鋰離子擴散阻抗和電荷轉(zhuǎn)移阻抗,從而提高材料的電化學性能[15]。 材料顆粒呈球形的原因在于添加的表面活性劑SDBS 具有一定的形貌導向作用,在表面活性劑SDBS 作用下,晶體在生長過程中逐漸呈現(xiàn)球形形貌。 這與韓夢瑩[16]的研究結(jié)論一致。

圖2 LiFePO4 材料SEM 照片
從圖2 還可以看出,產(chǎn)品無明顯團聚現(xiàn)象,粒度為微米級(1~15 μm)。 利用激光粒度儀進一步分析產(chǎn)品的粒度,結(jié)果見圖3。 結(jié)合圖2 和圖3 可知,合成 的LiFePO4材 料D10、D50、D90分 別 為1.93、6.34、13.26 μm,粒度分布范圍較窄。 這是由于,表面活性劑SDBS 的添加降低了產(chǎn)品的團聚性, 同時表面活性劑的合理添加能在一定程度上阻止樣品進一步長大[16-17]。粒度細小有利于縮短鋰離子的擴散距離,從而提升LiFePO4材料的倍率性能及循環(huán)性能, 減少樣品性能的衰減率[11-12]。
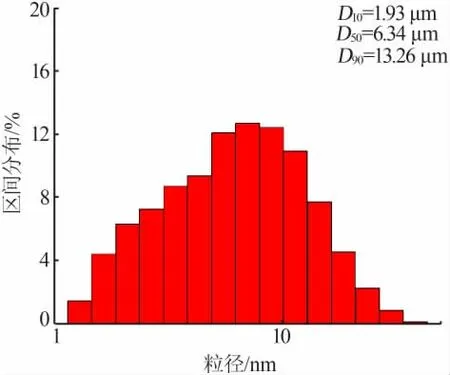
圖3 LiFePO4 樣品粒度分布圖
2.3 產(chǎn)品的電化學性能分析
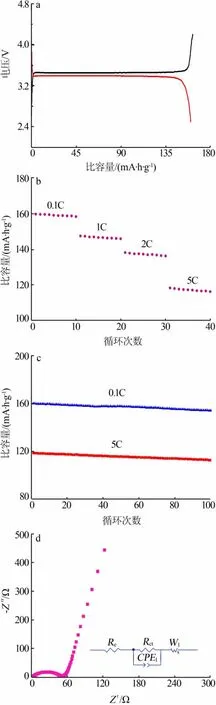
圖4 LiFePO4 材料的電化學性能
圖4a、b、c、d 為合成的LiFePO4材料首次充放電性能、倍率性能、循環(huán)性能和交流阻抗性能。 圖4a為LiFePO4材料0.1C 首次充、放電性能。 由圖4a 可知,LiFePO4材料在3.4 V 左右有穩(wěn)定的充電平臺,在3.3 V 左右有穩(wěn)定的放電平臺,兩者之間的差值較小,并且充放電平臺長而平坦。從圖4a 還可知,在0.1C 倍率下首次充放電比容量分別達到162.0、159.9 mA·h/g,庫倫效率達到98.7%,說明材料不可逆容量小[11],展現(xiàn)了較好的電化學性能。這主要是因為,Li+對Fe2+位的占據(jù)提高了鋰離子的擴散速率[13],同時球形形貌也提供了高的比表面積, 增強了電化學反應(yīng)的活性, 提高了材料的首次充放電性能和庫倫效率[15]。
圖4b 為LiFePO4材料的倍率性能圖,測試了不同倍率條件下材料放電比容量的衰減情況。 由圖4b可知, 在0.1C、1C、2C、5C 倍率下分別循環(huán)10 次,LiFePO4材料的比容量衰減并不明顯, 平均值分別為159.18、146.73、137.25、117.29 mA·h/g,其倍率性能(以1C/0.1C 保持率計)達到92.3%。
圖4c 為LiFePO4材料的循環(huán)性能圖,分別測試了在0.1C、5C 倍率下材料循環(huán)50 次和100 次的穩(wěn)定性情況。 由圖4c 可知,0.1C 倍率下循環(huán)50 次和100 次時, 材料的比容量保持率分別為98.7%和96.4%;在5C 倍率下循環(huán)50 次和100 次時,材料的比容量保持率分別為97.6%和95.2%。 實驗結(jié)果顯示,材料在低倍率(0.1C)、高倍率(5C)條件下都具有優(yōu)異的循環(huán)性能。
圖4d 為LiFePO4材料循環(huán)1 次的交流阻抗譜圖。 由圖4d 可知,材料的交流阻抗譜圖由高頻區(qū)的一個壓縮半圓和低頻區(qū)的一條傾斜直線組成。 研究發(fā)現(xiàn), 磷酸鐵鋰鋰離子電池在放電過程中,Li+從液態(tài)電解質(zhì)內(nèi)部向電極遷移, 先通過電極-電解液界面膜到達固體-電極界面,在界面處發(fā)生電荷遷移,最后Li+由固體表面向內(nèi)部擴散,充電過程為其逆過程[18]。 圖4d 中,位于高頻區(qū)的半圓對應(yīng)于電化學反應(yīng)中電荷的轉(zhuǎn)移過程,低頻區(qū)的斜線對應(yīng)于Li+在材料體相中的擴散過程。 因此對應(yīng)畫出了其等效電路(圖4d 插圖), 其中Re表示電解液電阻,Rct表示電荷傳遞電阻,W1為Li+在活性材料中擴散所產(chǎn)生的Warburg 阻抗,等效電路中常相位元件CPE1代表界面雙層電容參數(shù)。 根據(jù)等效電路圖擬合后的結(jié)果可知,Rct約為50.6 Ω,具有較低的電荷轉(zhuǎn)移阻抗,這可能是由于材料具有規(guī)整的球形形貌導致的[15]。
3 結(jié)論
以七水合硫酸亞鐵、磷酸二氫銨、一水合氫氧化鋰為原料,通過添加SDBS 作為表面活性劑,采用液相水熱法合成技術(shù),一步合成了磷酸鐵鋰正極材料。研究結(jié)果表明: 合成的材料為非化學計量比的Li1.02Fe0.994PO4;為正交晶系橄欖石結(jié)構(gòu),空間群為Pnma;為微米級球形顆粒,D50為6.34 μm。 電化學性能測試結(jié)果表明,在0.1C 倍率下首次充放電比容量分別為162.0、159.9 mA·h/g, 庫倫效率達到98.7%、倍率性能(以1C/0.1C 保持率計)為92.3%,0.1C 倍率循環(huán)100 次容量保持率為96.4%, 展現(xiàn)出良好的電化學性能。
