CDK1 磷酸化修飾對驅動蛋白Kif18A 的ATP 酶活性的影響
2020-06-10 02:13:42蘇春玉周之春朱長軍
天津師范大學學報(自然科學版) 2020年3期
蘇春玉,周之春,朱長軍
(1.天津師范大學生命科學學院,天津300387;2.天津師范大學天津市動植物抗性重點實驗室,天津300387;3.天津師范大學分子細胞系統生物學重點實驗室,天津300387)
驅動蛋白分子(kinesins)是以細胞內微管為軌道的動力蛋白,與細胞內細胞器的移動、細胞有絲分裂及減數分裂、組織器官的生長發育以及神經元的發育及信號傳導等密切相關[1].Kif18A 是Kinesin-8 家族中的一員.研究表明,Kif18A 對有絲分裂過程中的紡錘體微管動力學和染色體振幅起著重要調控作用[2-3].與其他驅動蛋白分子相同,Kif18A 蛋白由3個功能域組成:N 端的馬達結構域(motor domain)、中央卷曲螺旋桿狀結構域(stalk domain)和C 端貨物分子結合結構域(tail domain).Kif18A 利用水解ATP 釋放的能量可以沿著微管向正極末端移動,由于其C 末端存在另一個微管結合位點,因此Kif18A 能夠穩定運動到微管正極末端并聚集,進而抑制微管的動態不穩定性[4-7],通過降低染色體的振蕩速率限制其振蕩幅度[8-10].有絲分裂關鍵蛋白激酶CDK1 可對Kif18A 的S674 和S684 氨基酸位點進行磷酸化修飾,使得Kif18A 無法到達動粒微管的末端并聚集,因此Kif18A 無法發揮其降低微管動態不穩定性的功能,使得動粒振幅增大[11].據推測這可能是因為CDK1 的磷酸化修飾影響了Kif18A 的ATP 酶活性,使Kif18A 無法到達微管正極末端.
本研究構建了Kif18A 突變體的桿狀病毒Kif18AAA(S674A/S684A)和Kif18A-EE(S674E/S684E),利用SF9 細胞表達這2 種桿狀病毒蛋白,通過純化獲得Kif18A-AA 和Kif18A-EE 蛋白,檢測其ATP 酶活性,進而分析CDK1 磷酸化修飾對Kif18A 蛋白ATP 酶活性的影響,為深入理解Kif18A 在細胞有絲分裂期的功能提供體外實驗依據.
1 材料與方法
1.1 材料
昆蟲細胞SF9,本實驗室自存;pFastBac-HTa、pEGFP-Flag-Kif18A-EE、pEGFP-Flag-Kif18A-AA 質粒為本實驗室前期構建;TransT1、DH10Bac 感受態細胞、Kif18A 抗體、Kif18A 磷酸化抗體均為實驗室自制.
1.2 試劑
ATPase/GTPase ELIPA Biochem Kit(Catalog number:BK051/52)、Tubulin 和488 Labeled Tubulin,美國Cytoskeleton 公司;瓊脂糖凝膠回收試劑盒,北京康為世紀生物科技有限公司;EcoRⅠ和SalⅠ工具酶、SuperFectin 試劑,美國Thermo Scientific 公司;T4 連接酶,日本Takara 公司;SIM SF 培養基,北京義翹神州生物技術有限公司;鼠抗Flag 抗體、Flag Beads 和3×Flag Peptide,美國Sigma 公司.
1.3 方法
1.3.1 DH10Bac 感受態細胞轉化
從-80 ℃冰箱中取出DH10Bac 感受態細胞,冰上融化后,加入10 ng pFastBac-Flag-Kif18A-EE 或pFast-Bac-Flag-Kif18A-AA 質粒,冰上孵育30 min 后,42 ℃水浴鍋中熱激45 s,迅速置于冰上2 min,加入900 μL SOC液體培養基.37 ℃條件下225 r/min 搖菌6 h,之后取100 μL 涂平板(50 μg/mL Kan+,7 μg/mL Gen+,10 μg/mL Tet+,0.1 mol/L IPTG,2%X-gal),將平板倒置放于37 ℃培養箱中培養72 h.挑取3個白色克隆轉接到LB 液體培養基(含50 μg/mL Kan+、7 μg/mL Gen+和10 μg/mL Tet+)中,37 ℃過夜培養,提取桿狀病毒基因組.
1.3.2 桿狀病毒基因組提取
將菌液轉移到1.5 mL 離心管中,14000 r/min 離心1 min,真空吸干上清,加入0.3 mL 的Buffer P1(含有RNase A)重懸沉淀,混勻后加入0.3 mL 的Buffer P2,室溫孵育5min.加入0.3mL 醋酸鉀(濃度為3 mol/L,pH 值為5.5),輕輕混勻,蛋白形成白色沉淀. 冰上放置10 min后14000 r/min 離心10 min,將上清轉移到含有0.8 mL 異丙醇的離心管中.混勻,冰上放置10 min.室溫下14000 r/min 離心15 min,棄去上清后加入0.5 mL 70%的乙醇,室溫下14000 r/min 離心5 min.室溫放置10 min 風干沉淀,加入40 μL pH 值為8.0 的1×TE Buffer 重新溶解DNA 沉淀.-20 ℃儲存.
1.3.3 PCR 驗證桿狀病毒基因組
1.3.2 中得到的桿狀病毒基因組須進行PCR 驗證,確定其是否為發生轉座的重組基因組. 引物序列:M13F(5′-CCCAGTCACGACGTTGTAAAACG-3′),Kif18A-R(5′-CTAATGCCATCTCCCTTGAAAG-3′).PCR體系:93 ℃預變性3 min;94 ℃變性45 s,55 ℃退火45 s,72 ℃延伸5 min,30個循環;72 ℃終延伸7 min,4 ℃保存. 取5 μL PCR 產物進行瓊脂糖凝膠電泳,110 V 恒壓45 min.
1.3.4 重組桿狀病毒基因組感染SF9 細胞
在60 cm 平板中接種3×106個SF9 細胞,27 ℃下培養30 min,使細胞貼壁生長.吸棄培養基,加入3 mL新鮮培養基,加入轉染試劑(1 μg 病毒基因組DNA,15 μL SuperFectin 轉染試劑),邊滴加邊搖勻,27 ℃條件下培養72 h.
1.3.5 桿狀病毒的收取、擴增及滴度的測定
轉染72 h 后,收集上清為第一代桿狀病毒,避光保存在4 ℃冰箱中.由于轉染所得到的病毒滴度較低,不能高效表達目的蛋白,因此要進行桿狀病毒擴增,以增加病毒的滴度.用第一代桿狀病毒分別在10 cm平皿和50 mL 培養瓶中繼續感染SF9 細胞,擴增桿狀病毒,使其滴度逐步提高. 但過高的感染濃度可能會產生不完整的目的蛋白,因此通過調整感染比例(1 ∶100、1 ∶500、1 ∶1000、1 ∶5000)和感染時間(36、48、60、72 h)得到病毒表達蛋白的最佳條件.
1.3.6 Kif18A 突變體蛋白的表達和純化
在50 mL 錐形瓶中接種SF9 細胞,細胞密度為1×106mL-1,以桿狀病毒最佳感染濃度感染SF9 細胞,27 ℃條件下懸浮培養,在病毒表達蛋白的最佳時間收取細胞并將細胞沉淀凍存于-80 ℃冰箱中.
由于Kif18A 突變體蛋白在SF9 細胞中進行表達,因此蛋白要進行純化. 通過對融合到目的蛋白中的Flag 標簽蛋白進行純化,就可以得到目的蛋白.向50 mL SF9 細胞沉淀中加入2 mL 裂解液(0.3 mol/L NaCl,20 mmol/L pH 值為7.5 的Tris-Cl,1%NP-40,1 mmol/L PMSF,1 mmol/L DTT,10 mmol/L β-ME,0.5 mmol/L ATPNa,1μg/mL Approtinin,1×Cocktail,50 μmol/L Na3VO4),將細胞沉淀重懸后,冰上裂解60 min,將液體轉移至2 mL 離心管中.4 ℃下14000 r/min 離心30 min,轉移上清到2 mL 離心管中. 加入25 μL Flag Beads,4 ℃下旋轉孵育2 h 后收集上清,用洗液(0.3 mol/L NaCl,20 mmol/L pH 值為7.5 的Tris-Cl,1 mmol/L PMSF,1 mmol/L DTT,10 mmol/L β-ME,0.5 mmol/L ATP-Na,1μg/mL Approtinin,1×Cocktail,50 μmol/L Na3VO4)清洗Beads 3 次,加入洗脫液(3×Flag Peptide,稀釋比例為1 ∶20)4 ℃下洗脫2 h,收集第1 次洗脫樣品.再次加入洗脫液,4 ℃下過夜,洗脫后收集第2 次洗脫樣品.將2 次所得的純化蛋白進行聚丙烯酰胺凝膠電泳,考馬斯亮藍染色1 h 后進行脫色,觀察蛋白純化的效果.
1.3.7 體外微管合成
根據微管合成試劑盒(Cat.#TL488M)說明書合成微管. 取1 份分裝好的微管蛋白(Tubulin 和488 Labeled Tubulin 體積比為5 ∶1),迅速加入等量General Tubulin Buffer(80 mmol/L PIPES,2 mmol/L MgCl2,0.5 mmol/L EGTA,1 mmol/L GTP,1 mmol/L DTT,20 μmol/L Taxol).37 ℃水浴合成2 h 后,取0.5 μL 滴片,鏡下觀察微管合成情況,室溫保存合成的微管.
1.3.8 Kif18A 突變體蛋白的ATP 酶活性測定
首先根據ATPase/GTPase ELIPA Biochem Kit 說明書繪制磷酸根標準曲線. 將1mL ELIPA 反應溶液、12.5 μL ELIPA 試劑2、240 μL ELIPA 試劑1、80 μL合成微管和10 μL Taxol(2 mmol/L)混和后,按每孔147.5 μL 混合液的量在96 孔板中分裝,向每孔中加入1 μg 純化的Kif18A-EE 或Kif18A-AA 蛋白,每種蛋白設置3個重復. 以不加目的蛋白為空白對照,用洗脫液補齊.設置好分光光度計參數,在加入ATP 前預先測定2 組吸光度值.用排槍移液器同時向混合液中加入10 μL ATP 溶液,立刻置于分光光度計中測定不同孔的360 nm 處的吸光度值(OD360),每1 min 測1 次,連續測量1 h.處理數據,繪制ATP 酶活性曲線.
1.4 數據處理
將測得的3 組OD360取平均值,用目的蛋白的OD360減去空白對照的OD360,根據磷酸根標準曲線計算出差值對應的磷酸根的物質的量.用加入ATP 后的磷酸根物質的量減去加入ATP 前的數值,得到目的蛋白水解ATP 產生的磷酸根物質的量,分別根據2 種蛋白對應的磷酸根物質的量繪制ATP 酶活性曲線.
2 結果與分析
2.1 質粒pFastBac-Kif18A-EE 和pFastBac-Kif18AAA 的構建與鑒定
用EcoRⅠ和SalⅠ雙酶切pEGFP-Flag-Kif18AEE、pEGFP-Flag-Kif18A-AA、pFastBac-HTa 質粒后進行瓊脂糖凝膠電泳,結果如圖1 所示.pEGFP 空質粒大約為4800 bp,Kif18A cDNA 長度為2697 bp,因此電泳前面的條帶為Flag-Kif18A-EE 和Flag-Kif18A-AA.pFastBac-HTa 質粒長度為4855 bp,與DNA marker 進行比對,位置正確.
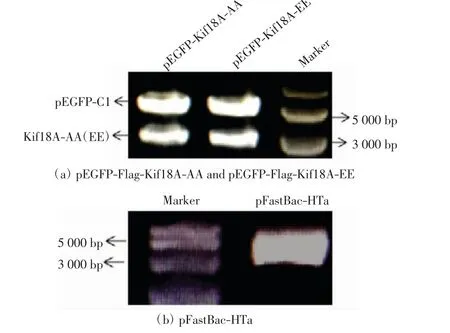
圖1 酶切3 種質粒后的瓊脂糖凝膠電泳檢測Fig.1 Electrophoresis of agarose gel of three kinds of plasmid after digestion
通過瓊脂糖凝膠回收試劑盒得到Flag-Kif18AEE、Flag-Kif18A-AA 和pFastBac-HTa 的線性DNA,電泳結果如圖2 所示.

圖2 膠回收后3 種質粒的瓊脂糖凝膠電泳圖Fig.2 Electrophoresis of agarose gel of three kinds of plasmid after gel extraction
通過T4 連接酶16 ℃過夜連接,連接產物全部轉化TransT1 感受態細胞,過夜培養,挑取陽性克隆,液體培養. 通過小提質粒試劑盒提取pFastBac-Kif18AEE、pFastBac-Kif18A-AA 質粒. 質粒經過測序后與NCBI 公布的序列進行比對驗證.
2.2 桿狀病毒基因組的制備及驗證
將pFastBac-Kif18A-EE、pFastBac-Kif18A-AA 質粒分別轉化至DH10 Bac 感受態細胞中,挑取白色克隆過夜培養,提取桿狀病毒基因組.當目的基因插入到桿狀病毒基因組中,如圖3 所示,會得到含有目的基因的重組桿狀病毒基因組. 使用轉座位點上游存在的M13 上游引物和目的片段中設計的下游引物進行PCR,可得到大小為3707 bp 的DNA 片段.對PCR 產物進行瓊脂糖凝膠電泳,條帶大小與理論值相符,證明確實發生了轉座,獲得了重組桿狀病毒的基因組.
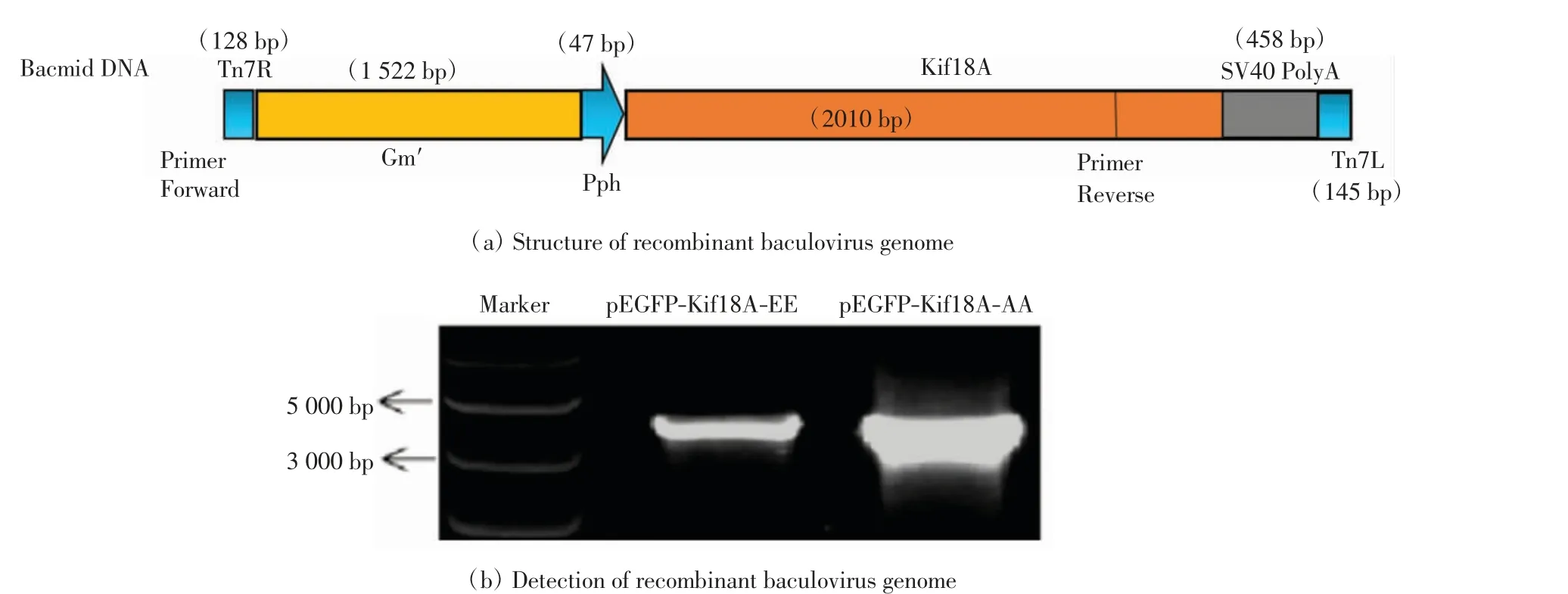
圖3 重組桿狀病毒基因組的驗證Fig.3 Verification of recombinant baculovirus genome
2.3 桿狀病毒的擴增及蛋白表達條件優化
將重組的桿狀病毒基因組轉染SF9 細胞后,桿狀病毒在SF9 細胞中進行組裝和增殖.當SF9 細胞中的病毒過多時,桿狀病毒就會從SF9 細胞中釋放,進一步感染其他的SF9 細胞.但病毒擴增到一定程度后,可能會表達不完整的Kif18A 蛋白,因此需要優化Kif18A突變體蛋白表達的病毒感染條件.以Kif18A-EE 桿狀病毒為例進行優化過程的描述.通過設置連續梯度的感染比例(1 ∶100,1 ∶500,1 ∶1000,1 ∶5000)感染SF9細胞進行蛋白表達,在表達時間(72 h)一致的情況下,可以得到桿狀病毒的最佳感染比例為1 ∶5000,如圖4(a)所示,在此感染比例下未出現雜蛋白. 如果無論何種感染比例都出現很多雜蛋白,則用1 ∶5000 的感染比例感染SF9 細胞,分別在36、48、60、72 h 收取細胞,通過Weatern Blot 驗證,得到蛋白的最佳表達時間為72 h,如圖4(b)所示.最終得到Kif18A-AA 的最佳感染比例為1 ∶20000,Kif18A-EE 的最佳感染比例為1 ∶5000,最佳表達時間均為72 h.
2.4 蛋白質的表達及純化
按照2 種桿狀病毒表達目的蛋白的最優條件感染SF9 細胞,收取細胞后通過蛋白印跡雜交實驗驗證所感染的SF9 細胞大量表達了目的蛋白,結果如圖5所示.

圖4 Kif18A-EE 桿狀病毒條件優化Fig.4 Condition optimization of Kif18A-EE baculovirus
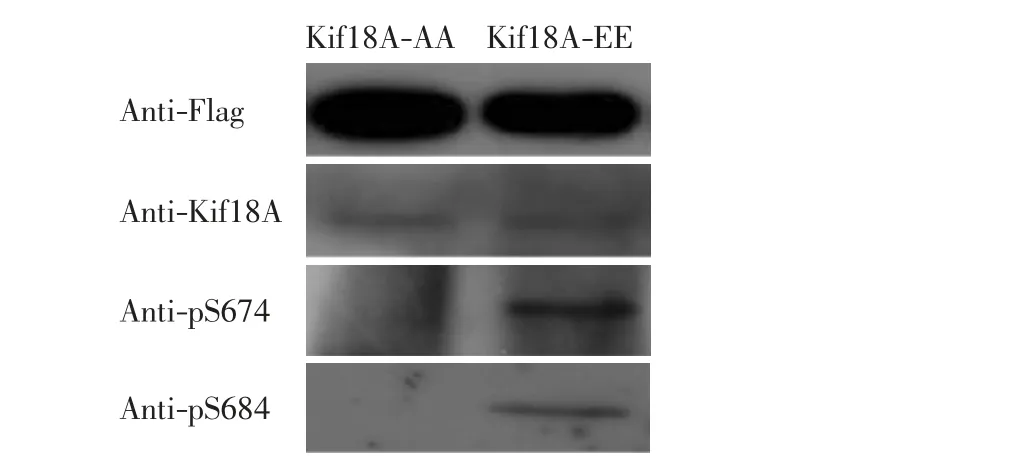
圖5 桿狀病毒表達蛋白的鑒定Fig.5 Identification of baculovirus-expressed proteins
由圖5 可以看出,SF9 細胞表達的蛋白可以被特異性鼠抗Flag 和兔抗Kif18A 抗體識別,Kif18A-EE 蛋白可以被特異性抗Kif18A-S674 和S684 位點磷酸化的抗體所識別,Kif18A-AA 蛋白不能被這些磷酸化抗體識別.表明Kif18A-EE 蛋白可以很好地模擬磷酸化的Kif18A 蛋白,而Kif18A-AA 蛋白模擬了非磷酸化的Kif18A 蛋白.
隨后利用Flag Beads 純化上述蛋白,純化后進行聚丙烯酰胺凝膠電泳,考馬斯亮藍染色后脫色,觀察目的蛋白的純化效果,結果如圖6 所示.純化得到的Kif18A-EE 和Kif18A-AA 蛋白濃度分別為0.12 μg/μL和0.10 μg/μL.
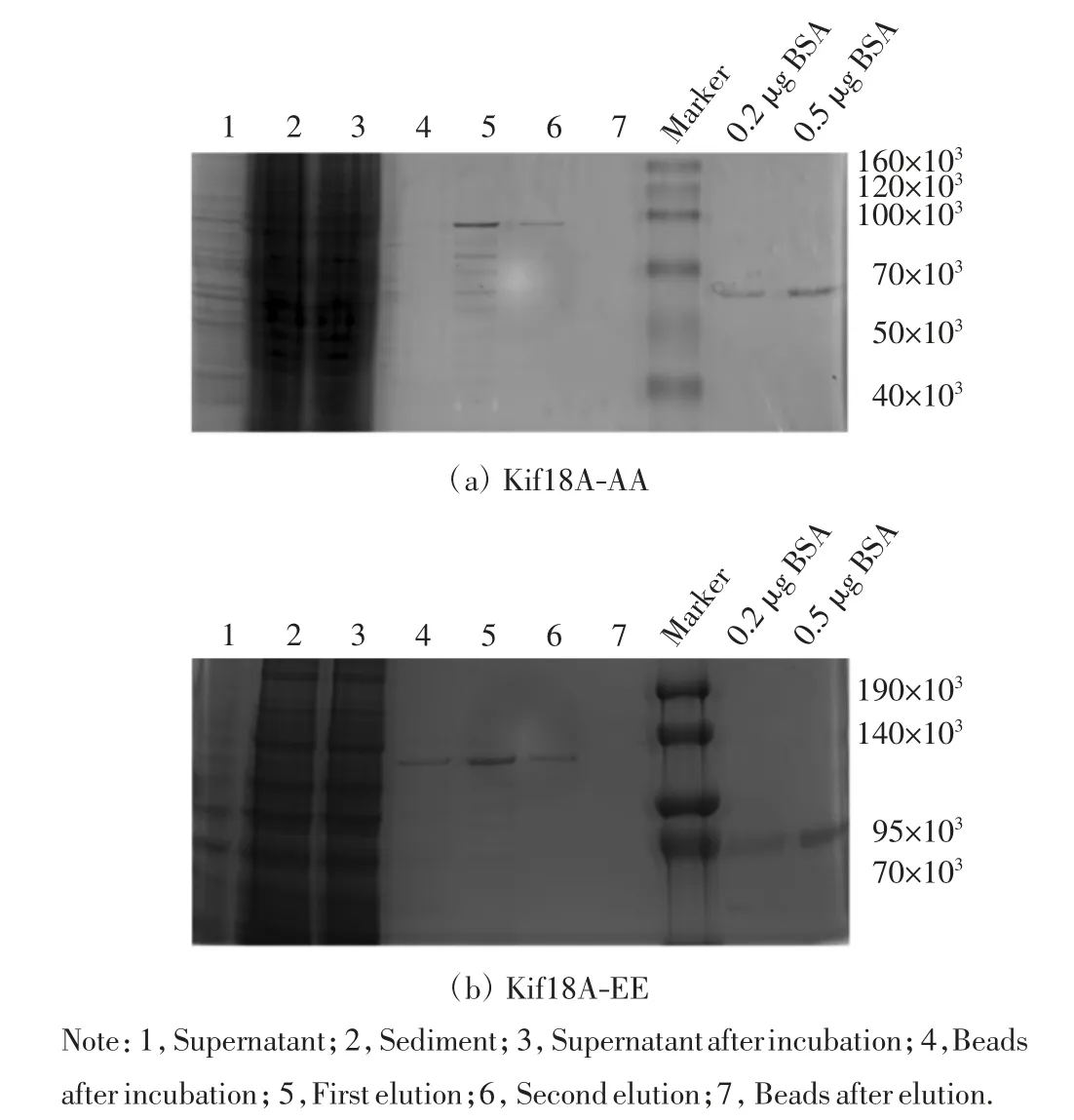
圖6 Kif18A-AA 和Kif18A-EE 的蛋白純化Fig.6 Purification of Kif18A-AA and Kif18A-EE
2.5 微管體外合成
Kif18A 作為一種驅動蛋白,能夠利用水解ATP 釋放的能量沿著微管向正極末端移動. Kif18A 要發揮ATP 酶活性,需要微管參與,因此必須進行體外微管合成.微管蛋白在GTP 存在下傾向于聚合,但聚合的微管并不穩定,所以在合成微管的過程中加入Taxol.微管蛋白通過聚合,合成了相對獨立的微管,如圖7所示,在熒光顯微鏡下觀察,可以看到發出綠色熒光的微管.
2.6 ATP 酶活性檢測
Kif18A 沿微管向正極移動會水解ATP,形成ADP和磷酸根,且每水解一分子ATP 會生成一分子磷酸根,因此通過磷酸根的量可以得出被水解ATP 的量,即被水解的ATP 的量反映Kif18A 的ATP 酶活性.磷酸根在360 nm 處有自己的吸收峰,因此可以利用分光光度計測量360 nm 處的吸光度值從而得到磷酸根的量.通過磷酸根的物質的量及其對應的吸光度值,得到磷酸根的標準曲線,如圖8 所示.

圖7 微管體外合成Fig.7 Microtubule synthesis in vitro
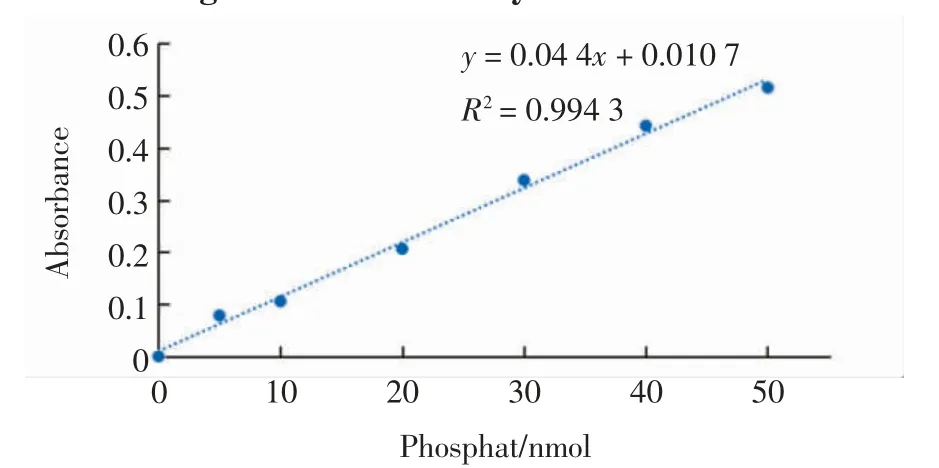
圖8 磷酸根標準曲線Fig.8 Standard curve of phosphate
利用純化得到的目的蛋白和體外合成的微管,進行ATP 酶活性檢測.根據加入目的蛋白后磷酸根的吸光度值,由磷酸根標準曲線計算出Kif18A 突變體蛋白水解ATP 的量,得到Kif18A 突變體蛋白的ATP 酶活性,結果如圖9 所示.
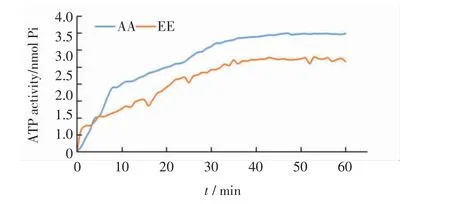
圖9 Kif18A-AA 和Kif18A-EE 的ATP 酶活性檢測Fig.9 Detection of ATPase activities of Kif18A-AA and Kif18A-EE
由圖9 可以看出,非磷酸化的Kif18A(Kif18A-AA)的ATP 酶活性進入平臺期后數值均為3.47 nmol Pi,模擬CDK1 磷酸化后Kif18A(Kif18A-EE)的ATP 酶活性平臺期數值約為2.72 nmol Pi,即Kif18A-EE 蛋白的ATP 酶活性低于Kif18A-AA 蛋白的ATP 酶活性,表明CDK1 對Kif18A 的磷酸化修飾降低了Kif18A 的ATP酶活性.
3 討論與結論
Kinesin-8 家族在整個真核細胞有絲分裂過程中對于微管長度的控制具有重要作用. Kif18A 作為Kinesin-8 家族中的一員,在微管正極末端的作用存在爭議:體外實驗發現Kif18A 能夠作為微管解聚酶使微管的穩定結構解聚[12],但也有研究表明,Kif18A 不具有內在的解聚酶活性,而是通過阻止微管蛋白加到微管末端,進而阻止了微管的聚合和解聚[2].Kif18A 對細胞周期中的染色體整列有很大的影響.染色體整列包括中板集合與形成最薄中期板(thinnest metaphase plate)2個過程.有研究[11]報道,Kif18A 在細胞有絲分裂早期被有絲分裂蛋白激酶CDK1 磷酸化,這種修飾可以被磷酸酶PP1 去磷酸化,當Kif18A 無法被PP1 去磷酸化時,細胞有絲分裂過程就無法形成最薄中期板.這可能是因為磷酸化的Kif18A 無法到達微管正極末端,從而無法降低微管的動態不穩定性.同時Kif18A 被磷酸化后不影響中板集合的時間,但會延遲最薄中期板形成的時間,這可能是因為CDK1 的磷酸化修飾影響了Kif18A 的運動能力.雖然H?fner 等[11]應用細菌表達的Kif18A 進行了ATP 酶活性的測定,但是由于原核細胞表達的蛋白可能不具有正常的蛋白構象[13],因而實驗結果仍需進一步驗證.本研究利用桿狀病毒昆蟲細胞表達系統獲得了目的蛋白,這種真核表達系統能夠使目的蛋白的免疫原性、ATP 酶活性等與天然蛋白的生物活性基本相同.體外實驗結果顯示Kif18A-EE 蛋白的ATP 酶活性低于Kif18A-AA 蛋白的ATP 酶活性,表明CDK1 對Kif18A 的磷酸化修飾降低了它的ATP 酶活性.這一結果預示著CDK1 的磷酸化修飾作用通過影響Kif18A 的ATP 酶活性,進而影響其在微管上的運動能力,使其無法及時到達微管末端并控制最薄中期板形成的時間. CDK1 磷酸化修飾調節Kif18A 的ATP 酶活性的機制仍不清楚,是通過影響Kif18A 的結構還是與其他蛋白的結合能力,仍需繼續深入探索.
