Effects of group IIIA element impurities on the visible light absorption of doped zinc blende ZnS
2020-05-13 13:49:18DONGMingHuiYUANGuangMingTANGShunLeiLIXiaoJie
原子與分子物理學(xué)報(bào) 2020年3期
DONG Ming-Hui, YUAN Guang-Ming, TANG Shun-Lei, LI Xiao-Jie
(Qilu Institute of Technology, Jinan 250200, China)
Abstract: ZnS has been used for splitting water to produce hydrogen, however, the reaction cannot be driven by the visible light because of its wide energy band gap.In this paper, by using density functional theory (DFT), the effects of group IIIA element impurities on the electronic structures and visible light absorption of doped zinc blende ZnS were studied.The results reveal that the structures of B, Al, Ga and In substituting for Zn are easy to form, while that of Tl substituting for Zn is not.Moreover, the absorptions of the doped structures in the visible light range are significantly enhanced, and their band edge positions are still suitable for water splitting to generate hydrogen, which means that B, Al, Ga and In substituting for Zn are candidate materials for water splitting driven by visible light.
Key words: Density functional theory (DFT); Absorption Coefficient; ZnS; Density of state
1 Introduction
With the development of technology andsociety, energy shortages and environmental pollution have become the major sectors restricting the country’s economic development and progress.Therefore, in order to ensure the development of the economy and society, it is urgent to find advanced technologies for producing renewable clean energy[1].Hydrogen (H2), as a clean, cheap, renewable energy, has attracted more attention[2, 3].Photocatalytic water splitting under sun light is an useful way for the generation of H2because it can convert light energy to hydrogen energy[4, 5].
In the hydrogen production procedure, ZnS has been used to splitting water to produce hydrogen[6].ZnS has two allotropic forms: a hexagonal form with wurtzite structure and a cubic form with zinc blende structure[7].At room temperature, the zinc blende structure is much more stable than the wurtzite one, and has high optical transmittance, wide energy gap (3.68 eV), high refractive index (2.35), and luminescence emissions from the ultraviolet to the visible spectrum[8].However, the wide band energy gap makes it cannot be effectively driven by solar light.Therefore, decreasing the band gap and increasing the light absorption are very important for the development of photocatalysts.Experiments and theory reveal that doping is an efficient method to tune the band gap (Eg) of semiconductors and facilitates excitation of electrons from valence band (VB) to conduction band (CB).By now, several metallic elements have been doped into ZnS to modify its optical properties.For instance, Sahraeietal.[9]indicated that Ni2+ions could decrease the optical transmittance.Sharmaetal.[3]reported that Zr could reduce the band gap of ZnS from 3.7 eV to 2.9 eV.Alvarez-Coronadoetal.[8]observed a red shift of the optical absorption edge after Cu and Mn ions doped in ZnS.Azmand et al.[10]found that the band gap energy of undoped ZnS was estimated to be 3.93 eV, and reduced to 3.16 eV after Se-doping.Azmandetal.[11]clarified that Al-doping decreased the band gap energy of the ZnS samples from 3.93 to 3.50 eV, and Hurma[12]also found similar results in nanocrystalline ZnS and Al doped ZnS films.Gaikwad et al.[13]revealed that Pd doped ZnS showed obvious photocatalytic activity when irradiated with visible light compared to ZnS.Wangetal.[14]found Cu-N co-doped ZnS can reduce the band gap and enhance its photocatalytic properties.Density functional theory (DFT), especially the first-principle calculations, have proved to be invaluable in understanding the physical and chemical properties of nanomaterials.Further survey of literature reveals that DFT has been performed on the effect of carbon group elements[15], Si[16,17], N[18], H[19], V[20]doped ZnS on the optical properties.Recently, (B[21], Al[22,23], Ga[24,25], In[26])-doped ZnS has been successfully synthesized in experiment.Through Liu[27]has studied the dielectric function and absorption coefficient spectra of Al and Ga doped ZnS, however, other properties such as formation energy, band edge positions has not calculated.Therefore, in order to systematically understand the properties of group ⅢA elements doped ZnS, the formation energy, dielectric function, absorption coefficient spectra, band edge positions and density of states have been presented by using first principles based on DFT.
2 Computational details
All of the calculations were performed using first principle[28]based on DFT[29].Generalized gradient approximation (GGA) and the Perdew-Burke-Ernzerhof (PBE)[30]function were employed as exchange-correlation potential.The interaction between valence electron and ion core was ultrasoft pseudopotential.For PBE potential, only Zn(3d104s2), S (3s23p4), B(2s22p1), Al(3s23p1), Ga(3d104s24p1), In(4d105s25p1), Tl(5d106s26p1) are considered as the valence electron.The cut off energy was set as 400 eV and Monkhorst-Pack[31]k-points were chosen to 4×4×4 in the Brillouin zone to ensure that all of the calculations were converged.For the geometry optimization process, the energy change and the maximum tolerances of the force, stress, and displacement were set to 2.0×10-5eV/atom, 0.05 eV/?, 0.05 GPa and 0.002 ?, respectively.
3 Results and discussion
3.1 Geometry optimization results
The zinc blende ZnS possesses stable structure and good photocatalytic performance with a space group of 216 (F4-3m).The atomic position of Zn and S atoms are selected at (0, 0, 0) and (1/4, 1/4, 1/4), respectively.Fig.1 shows a unit cell structure of pristine ZnS, which was optimized by the first principles calculation.
The optimized cell parameters are shown in Table 1.It can be seen that the optimized lattice parameters of pristine ZnS crystal cell area=b=c=5.425 ?, which are in good agreement with the experimental data (a=b=c=5.41 ?[32, 33]).Other previous theoretical research reports[15, 34]are also in good agreement with our results.These analyses show that our computational method is reasonable.
3.2 Impurity formation energy
The formation energy(Ef) is used to evaluate the
Table 1 The optimized cell parameters, crystal volume changes for the pristine, ZnS: B, ZnS: Al, ZnS: Ga, ZnS: In and ZnS: Tl, as well as experimental values.
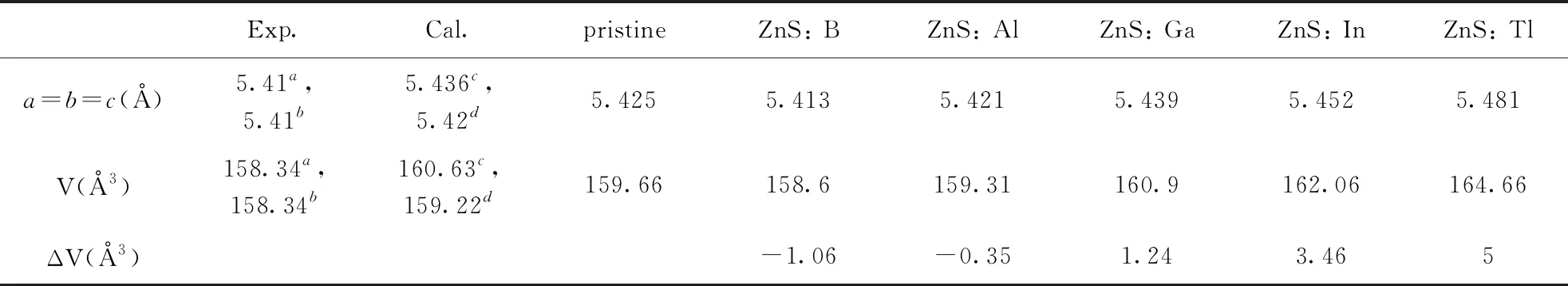
Exp.Cal.pristineZnS: BZnS: AlZnS: GaZnS: InZnS: Tla=b=c(?)5.41a,5.41b5.436c,5.42d5.4255.4135.4215.4395.4525.481V(?3)158.34a,158.34b160.63c,159.22d159.66158.6159.31160.9162.06164.66ΔV(?3)-1.06-0.351.243.465
aref.[32],bref.[33],cref.[15],dref.[34]
relative stability of a doping system and the degree of difficulty of atomic doping.The smaller theEfis, the more stable will be the structure.Formation energy can be calculated by using the following equation[35].
Ef=E(doped)-E(pristine)-μX+μZn
(1)
whereE(doped) andE(pristine) are total energies of doped ZnS and pristine ZnS, respectively.TheμX, andμZnstand for the chemical potentials of dopants atom and Zn, respectively.μZnis very close to each Zn atom energy in bulk Zn, and so doesμX.The formation energies of five doped systems are given in Fig.2.
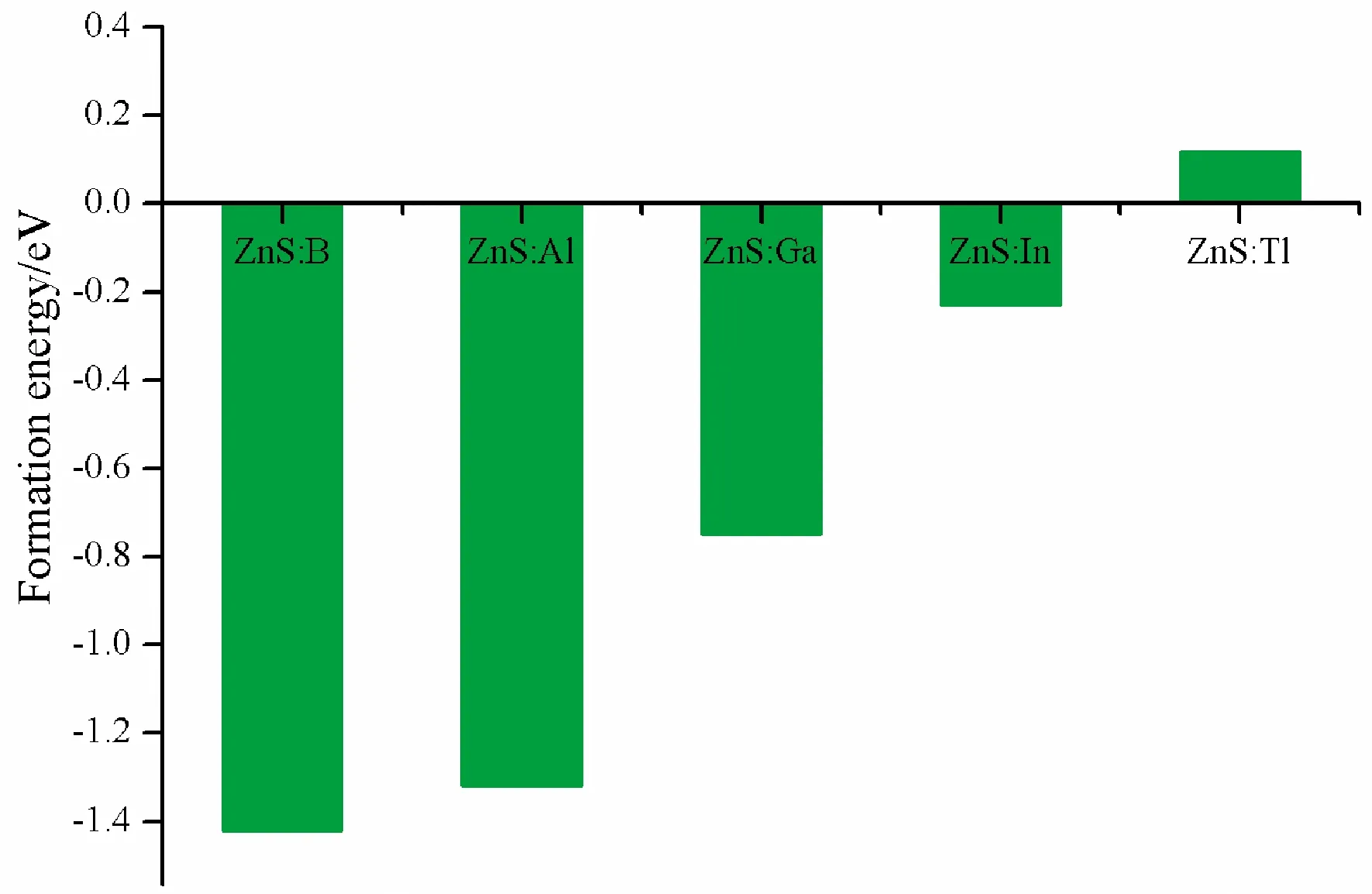
Fig.2 The formation energies of pristine and five doped systems
Fig.2 reveals that formation energies for structures of B, Al, Ga and In substituting for Zn are all negative, which imply that all structures are stable.However, for Tl substituting for Zn, the formation energy is 0.12 eV, which indicates that this structure is not easy to form.This is because the radius of Tl is larger than Zn, therefore, the lattice distortion caused by substitution is very serious.As shown in Table 1, the volume changes by B, Al, Ga and In substituting for Zn are about 1 ?3, however, for Tl is 5 ?3.Therefore, in the following work, the properties of B, Al, Ga and In doped ZnS systems has been studied.
3.3 Band structures
It must be mentioned that all of our calculations in this paper were based on the DFT.However, experience has proved that the energy gap calculated by DFT is smaller than that obtained by experiments[36].In order to overcome this defect, scissors operator is usually used to correct the results in optical properties[37, 38].In this paper, the energy gap of pristine ZnS is 2.93 eV, while the reported is 3.68 eV[8], therefore, 0.75 eV was chosen as scissors operator.Fig.3 shows that the calculated band gap of pristine ZnS, and B, Al, Ga, In substituting for Zn are 3.68 eV, 2.83 eV, 2.65 eV, 3.06 eV, 3.23 eV, respectively.All of the band gaps of impurities systems are narrower than that of pristine structure.This is beneficial for the transfer of the photogenerated electrons from valence band maximum (VBM) to the conduction band minimum (CBM), improving the solar light absorption and enhancing the photocatalytic properties of ZnS.
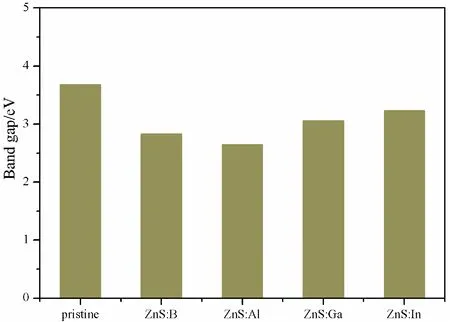
Fig.3 The band gap of pristine and various doped systems
3.4 Optical properties
In order to calculate theeffects of element impurities on optical properties, the dielectric function and absorption coefficient have been calculated.As we all know, the dielectric function can explained as the transitions between occupied and unoccupied states.In the linear response range, the optical material properties can be determined from the dielectric function[39, 40]
ε(ω)=ε1(ω)+iε2(ω)
(2)
The imaginary partε2(ω) of dielectric function determines the probability that electrons jump from the valence band to the conduction band.The bigger the imaginary part is, the more obvious the effect is[41].The imaginary partε2(ω) of pristine ZnS and element impurities are shown in Fig.4.It can be seen that the peaks of doped structures are all located at 2.51 eV, 2.23 eV, 2.72 eV and 2.83 eV, respectively, however, pristine structure is in the ultraviolet region.Therefore, the imaginary partε2(ω) edges of all doped structures shift towards the visible light region in comparison with the pristine one.This is consistent with the band gap change regulation.
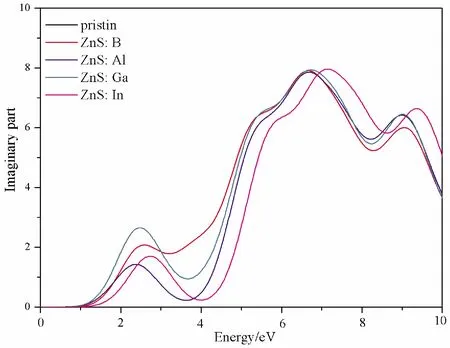
Fig.4 The imaginary part of the dielectric function of the pristine and doped ZnS systems.
Based on the real partε1(ω) and imaginary partε2(ω) of dielectric function, the optical absorption coefficient can be calculated through the following expression[42].
(3)
where,I(ω) is absorption coefficient.The absorption coefficient of pristine and doped ZnS are presented in Fig.5.
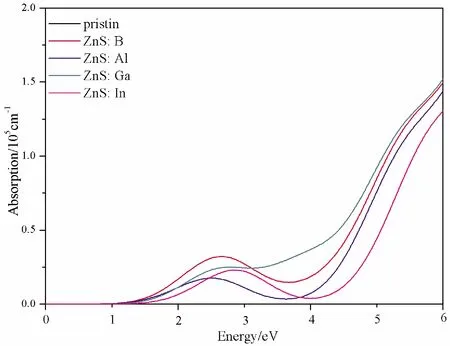
Fig.5 The absorption coefficient spectra of pristine and doped ZnS systems
For pristine ZnS, the obvious absorption peak occurs in the ultraviolet region, however, for the doped systems, the peaks of absorption coefficient appear in the visible light region.Therefore, compared with the pristine one, the absorption spectra shift towards the visible light region.This is consistent with the band gap and imaginary partε2(ω) change regulation in the visible light region.The results of calculation demonstrate that B, Al, Ga and In could significantly enhance the absorption coefficient of ZnS in the visible light range, especially for ZnS: B, ZnS: Ga and ZnS: In.
3.5 Hydrogen production
It is well known that the band edge positions of a semiconductor and the redox potentials of the adsorbate are very important to the photocatalytic performance.Thus, in this paper, the band edge positions of the conduction band and the valence band were calculated by the following expression[43].
(4)
EVB=ECB+Eg
(5)
where,EVBandECBare the VB and CB edges potentials, respectively.Xis the Mulliken electronegativity of the system, and theXis calculated to be about 5.26 eV for ZnS[44, 45].Eeis the normal hydrogen electrode (NHE) (~ 4.5 eV), andEgis band gap.Fig.6 shows the schematic diagrams for pristine and B, Al, Ga, In substituting for Zn as photocatalyst.In particular, conduction band potential for pristine and element impurities are -1.08 eV, -0.655 eV, -0.565 eV, -0.77 eV, -0.855 eV, respectively, while the valence band potential of these structures are 2.6 eV, 2.175 eV, 2.084 eV, 2.29 eV, 2.375 eV, respectively.According to the conditions of water splitting[44]: the valence band potential of photocatalyst should be higher than that of O2/H2O (1.23 eV vs NHE), and the conduction band potential should be lower than that of H+/H2O (0 eV vs.NHE).Fortunately, all the conduction band potentials are more negative than H+/H2(0 V) and hence can reduce H+to H2.On the other hand, the valence band potentials are all more positive than O2/H2O (1.23 V) and have the ability to oxidize H2O to produce O2.Among these doped structures, ZnS: Al has the lowest reducibility to produce H2and the lowest oxidation ability to produce O2, while opposite to the ZnS: In.
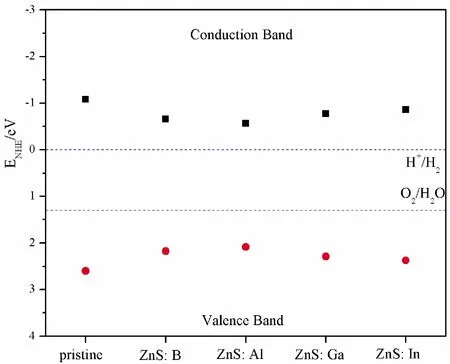
Fig.6 The calculated band potential of pristine and doped ZnS systems
3.6 Density of states (DOSs)
To further analyze the effect of substitutional elements on the electronic structure of ZnS, the density of states (DOSs) for the doped systems are successively shown in Fig.7.In each calculation, the highest occupied state is featured as the Fermi level (EF) and is set to zero, and he dotted lines at the zero point of horizontal coordinates represent the Fermi level.Fig.7(a) shows that the valence band maximum (VBM) of pristine ZnS is mainly dominated by S-3p and Zn-3p states, while the conduction band minimum (CBM) includes a small number of 3p, 4s states of Zn and 3s, 4p states of S.These states will result in a 3.68 eV energy band gap.For the doped structures, s states of the dopants have been found in the band gap.These impurity states cross the Fermi level serve as springboard under excitation, which are benefit for the transfer of the photogenerated electrons to the conduction band, improving the solar light absorption and utilization, and enhancing the photocatalytic properties of doped ZnS.Meanwhile, shallow donor levels above the CBM also act as capturing centers for photo-generated electrons, thus the recombination probabilities of electrons and holes will be reduced.On the other hand, doping will impel the valence band to move downward stronger than that of the conduction band, therefore, the energy band gap decreases to 2.83 eV, 2.65 eV, 3.06 eV and 3.23 eV, which might result in red shift of absorption spectra in experiments.The small energy band gap can significantly enhance the absorption in the visible light range, and the results have been confirmed in Fig.4 and Fig.5.
4 Conclusions
In summary, the electronic structures and optical properties of group ⅢA elements doped zinc blende ZnS are thoroughly studied based on the density functional theory.The obtained conclusions are as following.
(1)The analyses of formation energy reveal that the structures of B, Al, Ga and In substituting for Zn are easy to form, while that of Tl substituting for Zn is not.
(2)B, Al, Ga and In substituting for Zn on the one hand could narrow band gap obviously, which will enhance the solar light absorption significantly,on the other hand, it satisfy the requirement of water splitting to generate hydrogen.
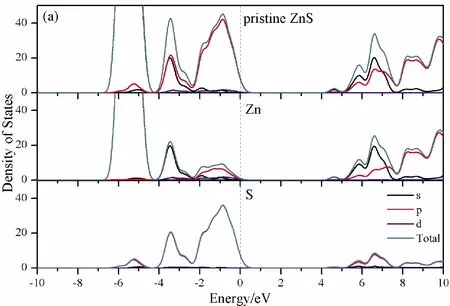
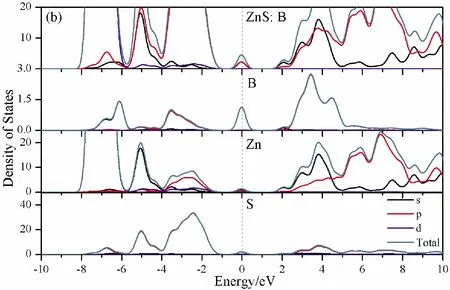
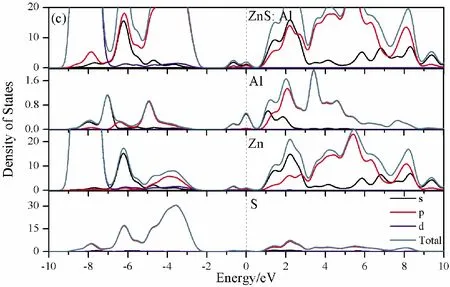
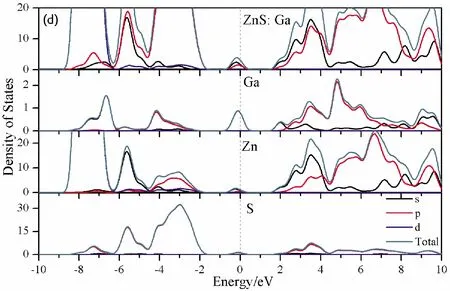
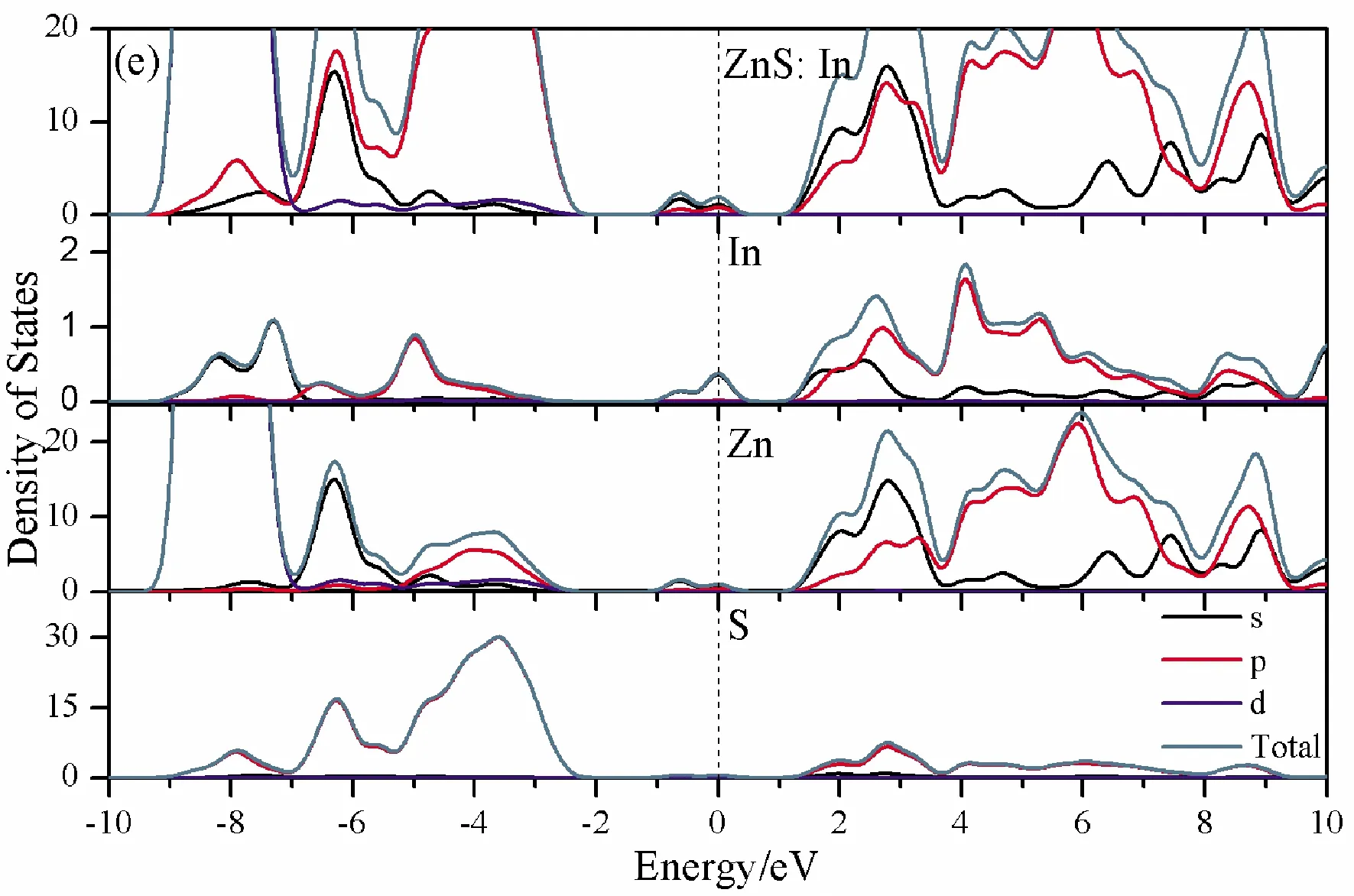
Fig.7 Density of states (DOSs) of (a)Pristine ZnS; (b)ZnS: B; (c) ZnS: Al; (d) ZnS: Ga; (e) ZnS: In

- 原子與分子物理學(xué)報(bào)的其它文章
- 原子尺寸對(duì)Lennard-Jones液體玻璃轉(zhuǎn)變過(guò)程中動(dòng)力學(xué)不均勻性的影響
- 尋找囚禁在光腔中的玻色-愛(ài)因斯坦凝聚體的八重準(zhǔn)晶
- Ab initio calculations of structural evolution and conductance of binary compound GaN chain on gold leads
- 稀土元素(La,Y)摻雜GaN的第一性原理計(jì)算
- 新型稀磁半導(dǎo)體Fe摻雜LiZnP的光電性質(zhì)
- La摻雜6H-SiC電子結(jié)構(gòu)和光學(xué)性質(zhì)的第一性原理研究