堿土金屬吸附二維α1硼烯結構和性質研究
2020-05-13 08:45:22韓健偉邊偉樾羅有華
原子與分子物理學報 2020年4期
韓健偉, 邊偉樾, 劉 錦, 徐 豪, 張 孟, 王 瀟, 羅有華
(華東理工大學,上海 200237)
1引 言
隨著2004年Novoselov等人微機械剝離制備單層石墨烯(graphene)的成功,以石墨烯為代表的新型二維晶體材料因其獨特的幾何結構和物理化學性質迅速成為了國內外的研究熱點[1].硼元素的電子結構為1s22s22p1,同時具有金屬和非金屬的雙重屬性,表現出獨特的成鍵方式.2007年,Tang等人通過理論計算發現了一種由三角形晶格和六角蜂窩狀孔洞兩者混合而成的新型α-sheet硼單層結構[2,3].2012年曾曉成課題組進一步通過CALYPSO程序計算提出了多種形式的二維硼原子層,并且預測了能量上穩定的α1和帶皺褶的α'型硼烯結構[4,5].隨后,國內外不同研究小組分別計算模擬了在Au(111)、Ag(111)以及 MgB2等襯底生長出單層硼烯的可能性,為實驗上的成功制備硼烯指出了方案[6-8].最近中美兩國科學家同時在金屬襯底上成功制備出了單層硼墨烯, 對將來可能的基于硼烯的潛在應用提供了誘人的前景[9,10].
在二維硼材料領域實驗和理論已經開展了很多有價值的工作,目前這些研究主要集中在硼平面材料的結構預測與分析、不同結構的穩定性以及電子性質等.而對二維硼烯的吸附特性以及金屬原子吸附硼烯后體系的穩定性、結構變化和電子結構等問題的研究鮮有報道.另一方面,通過吸附和摻雜外來原子等化學修飾方法是提高納米材料結構穩定性以及改變材料物理化學性質的重要手段[11-15].硼原子具有復雜的成鍵機制和多配位能力,同時由于缺電子特性,使得硼原子易于與具有給電子特性的金屬原子發生相互作用,形成金屬硼化物,因此可以通過吸附不同金屬的種類來實現調控硼烯的性質.之后譚心團隊細致地分析了堿金屬吸附在石墨烯和磷烯表面的吸附性質和遷移行為,發現堿金屬在兩種二維納米材料表面的最穩定吸附位是空位,并且隨著原子序數的增大體系的遷移行為越明顯[16-17].2014年,Banerjee等人使用密度泛函方法結合從頭算分子動力學模擬研究了α1硼烯吸附鋰原子的情況,發現鋰原子吸附在硼烯六角空位正上方在能量上是最穩定的,并進一步討論了硼烯作為鋰離子電池陽極材料的可行性[18].;隨后Zheng 等人利用密度泛函理論計算研究了堿金屬鋰、鈉和鉀原子吸附于α硼烯后體系的穩定性和功函數變化[19,20],目前已有的文獻集中在堿金屬吸附硼烯的研究情況.堿土金屬最外電子層上有兩個s價電子,容易失去電子,因而可以和缺電子的硼形成穩定的堿土金屬硼化物.據我們所知,目前還沒有相關的堿土金屬原子或團簇吸附硼烯的文獻報道.
本論文工作采用第一性原理對新型α1硼烯吸附堿土金屬原子(鈹、鎂和鈣)及其二聚體團簇后體系的幾何結構、最低吸附點位、穩定性以及相互作用進行系統地研究.探索堿土金屬原子及其二聚體對二維硼烯結構和穩定性的影響,找到最佳吸附位置,并進一步研究復合體的吸附能、電荷轉移、態密度等性質,揭示硼烯中硼原子與外來堿土金屬原子的成鍵模式,為今后新型二維硼烯材料的性能調控提供可靠的理論依據.
2計算模型和方法
本文采用基于第一性原理的密度泛函理論(DFT)方法對鈹、鎂、鈣三種堿土金屬原子及其二聚體團簇在硼烯表面的吸附性質進行研究.所使用的硼烯為理論上最穩定α1硼烯并且采用(3×3)的超晶胞進行模擬計算,計算軟件為Materials Studio 中的CASTEP模塊,α1硼烯的真空層設置為15 ?,截斷能設為190 eV,布里淵區的K點在Monhkorst-Pack方法計算下選為2×2×1,能量的收斂值為2.0e-5 eV/atom,最大的應力收斂值為0.05 eV/?,最大壓強收斂值為0.1 GPa,最大位移收斂值為0.002 ?,最大迭代次數為200次,并且使用廣義梯度近似GGA-PBE泛函對體系進行幾何結構優化,能量以及其它性質的計算.經研究發現堿土金屬原子吸附于硼烯的位置主要有以下四種:空心位、頂位、橋位以及三角位,并且硼烯中具有一定的對稱性,一個充滿的六邊形硼烯中擁有7個硼原子,考慮等價位之后有3中不同的硼原子B1、B5、B13[5,6],最后我們還列出了在吸附過程中遇到的兩種特殊吸附位Near B13以及Near Hollow,如圖1所示,為了比較不同體系的穩定性,本文定義了硼烯結構吸附能:Eb=EB+EAM-ET,其中,Eb為總吸附能,EB為純的硼烯的能量;EAM為孤立堿土金屬原子的能量;ET為硼烯吸附堿土金屬后復合體的總能量.吸附能若是負值表明這個體系時無法共存的,而吸附能的值為正的,則表明有相互作用.計算所得吸附能值越大,表明堿土金屬原子與硼烯結合作用越強;反之,表明二者的結合作用越弱.
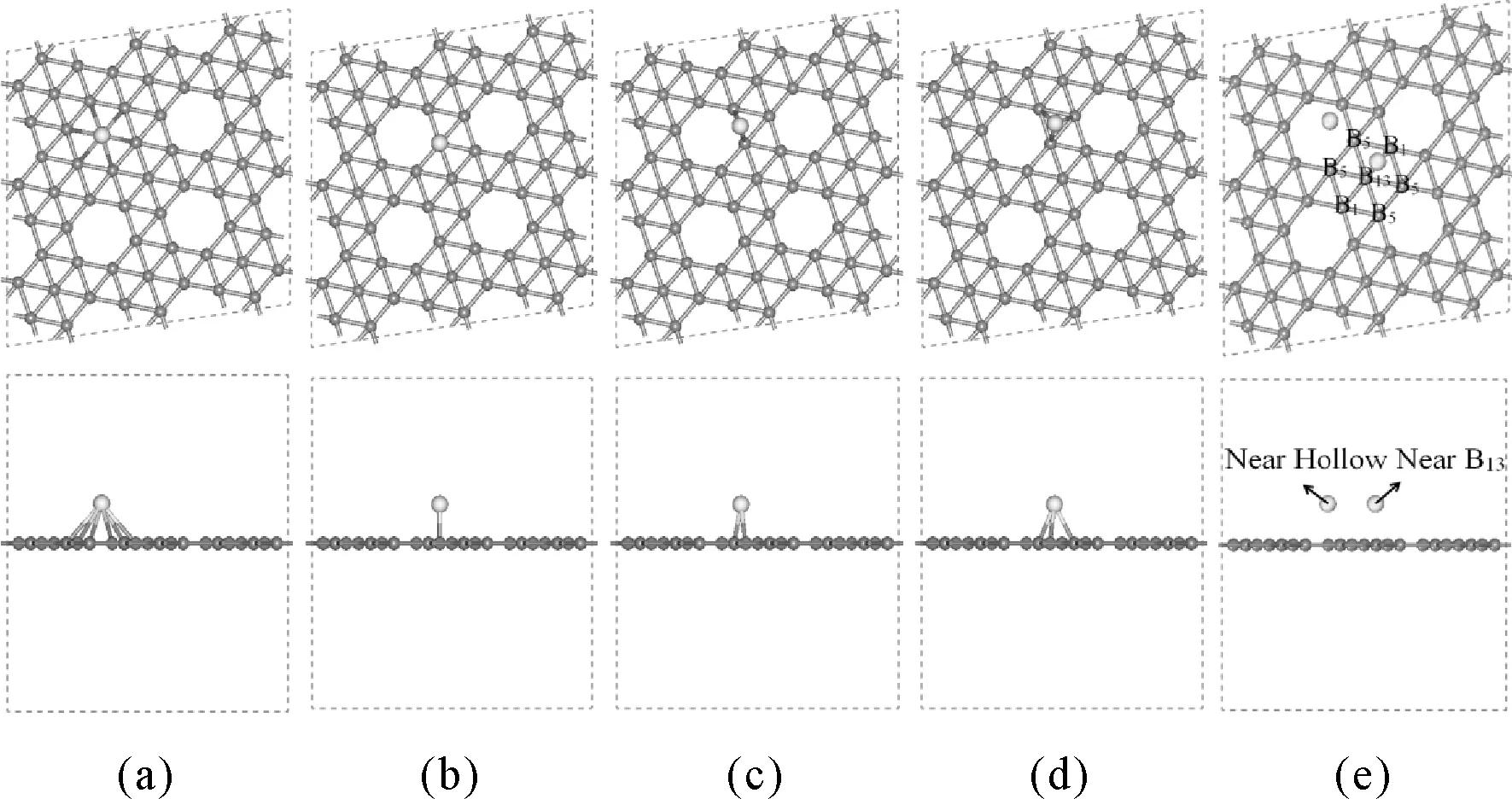
圖1 α1硼烯表面五個高對稱吸附位的俯視圖和側視圖. (a)空心位Hollow,(b)頂位Top,(c)橋位Bridge,(d)三角位Triangle,(e)Near B13以及Near HollowFig. 1 Top view and side view offive high symmetry adsorption sites of borophene. (a)Hollow,(b)Top,(c)Bridge,(d)Triangle,(e)Near B13 and Near Hollow
3結果和討論
3.1單個鈹、鎂、鈣原子在α1硼烯表面的吸附性質
由于α1硼烯是通過硼原子以三角形和六邊形混合的組合方式排列的,因此整個平面的對稱度是不如石墨烯的(所有的碳原子都是等價的),本工作選取擁有63個硼原子的(3×3)的超晶胞,其中有三種不同的硼原子,導致同一類高對稱吸附位就存在多個不同的吸附位置,為了便于比較,經過計算我們找到了每一類吸附位中吸附能最高的或吸附特征較為特殊的幾種,并就對這幾種不同的堿土金屬的不同吸附位中吸附能最大的一種進行比較,得到的堿土金屬—硼烯體系的能量和吸附能見表1和最穩態圖2.
從表1和圖2可以看出,在堿土金屬—硼烯的體系中,三種不同的堿土金屬Be、Mg和Ca無論在何種位置進行吸附體系的吸附能總是正的,表明堿土金屬與α1硼烯確鑿發生了相互作用,應當存在電荷之間的轉移或移動從而發生化學吸附.分析數據可以看到三種堿土金屬吸附在α1硼烯表面時的特征是具有較大區別的.對于鈹原子來說,當吸附的位置選擇除了Top(B13)這個位置,無論是其它的頂位,抑或是橋位、三角位、空心位,最終穩定的位置都是靠近原吸附位最近的一個空位,而在Top(B13)這個位置鈹原子會最終穩定在最初吸附的位置,可見鈹原子吸附在α1硼烯只有兩個穩定的位置,并且從吸附能可以看到當鈹原子穩定在空位時是最穩定的,Top(B13)只是一個亞穩態,兩者總能量相差2.025 eV左右.對于鎂原子來說吸附的位置全部與最初的試探位是相一致的,可見上文所提到的所有的可能的吸附位,對于鎂原子來說都是可以穩定的,并且在空位吸附時鎂原子具有最大的吸附能.而對于鈣原子來說情況稍顯復雜,總結一下可以知曉,鈣原子吸附在α1硼烯上大抵有三個較為穩定的位置,Near B13、Near Hollow以及空心位,具體吸附的位置與最初的試探位置有關,試探位靠近哪個位置,則最終就會吸附在那個位置,并且前兩個位置的吸附能差距并不是很大,Near B13與Near Hollow體系總能量相差0.03 eV,而單獨穩定在Hollow位時的吸附能是最小的.

表1 堿土金屬原子在α1硼烯表面不同吸附位的吸附能.其中,Site1為優化前初始試探位,Site2為優化后最終吸附位,ET為體系總能量,Eb為吸附能,△E為與最穩態的相對能量差

圖2 鈹、鎂、鈣原子吸附在α1硼烯上最穩態的俯視圖和側視圖,(a)、(b)和(c)分別對應鈹、鎂、鈣原子Fig. 2 The most stable top and side views of beryllium, magnesium and calcium atoms adsorbed on α1 borophene. (a), (b), and (c) correspond to beryllium, magnesium and calcium atoms, respectively.
值得注意的是在文獻19、20中,Li 原子處于α1硼烯不同位置的預設結構優化后均指向六邊形孔(Hollow位)中心上方的位置,與我們工作中Be和Mg的結果是一致的. 但Ca原子的最低能量吸附位置發生了改變,根據我們的計算,Near B13(非正上方)是最穩定的吸附位置.
比較三種堿土金屬吸附在α1硼烯表面的吸附能可以看到,鎂原子與α1硼烯的吸附能是最大的,鈣原子次之,而鈹原子雖然與α1硼烯存在相互作用,但吸附能約為3.7 eV,明顯小于Mg和Ca堿土金屬原子吸附的情況,但高于文獻17、18報道的單個Li原子的吸附能2.33 eV.另外我們測量了堿土金屬吸附在α1硼烯表面的距離,發現鈹原子與硼烯表面的距離為0.739 ?,鎂原子為1.991 ?,而鈣原子為2.532 ?,鈹原子與α1硼烯表面的距離過近,對α1硼烯表面的破壞程度是最大的,因此吸附能最小,鈣原子雖然具有較大的原子半徑,但它與硼烯表面的距離比鎂原子要大,這可能是鈣原子的吸附能略小于鎂原子的原因.綜上α1硼烯具有較強的儲鎂以及儲鈣的功能,由于鈹原子特殊的吸附位,α1硼烯在儲鈹方面也存在應用的空間,潛力較大.
3.2單個堿土金屬吸附在最穩處時的性質研究
在找到堿土金屬原子吸附α1硼烯的能量最低的吸附位置基礎上,通過計算所研究體系的Mulliken布局分析以及電子態密度圖來進一步研究α1硼烯吸附堿土金屬原子后體系的電子結構性質,分析其相互作用. 從Mulliken布局分析可以得到鈹原子,鎂原子和鈣原子在吸附后分別失去了1.35e,1.39e和1.30e電量的電荷,這些電子主要來自于最外層s軌道.對于鈹原子來說,鈹原子所失去的電子大都轉移到了六邊形孔洞上的4個B5原子和兩個B1原子上,4個B5原子各自獲得了0.21e,而兩個B1原子分別獲得了0.25e,可見鈹原子與硼烯中六邊形孔洞硼環的6個硼原子之間具有很強的等量電荷交換,鈹與整個硼烯體系的作用幾乎局限于六邊形孔洞附近.對于鎂原子,它所失去的電子則分散的相對較廣,其中六邊形孔洞上的4個B5原子各自獲得了0.23e,而兩個B1原子各自獲得了0.11e,并且鎂原子的部分電子轉移到了距離六邊形孔洞較遠的硼原子上,可見相對于鈹原子,鎂原子與整個α1硼烯表面的作用要更充分.鈣原子的穩定位不同于鈹、鎂原子,在吸附過程中轉移的電子大都轉移到了被充滿的六邊形上,6個硼原子各自平均獲得0.12e,并且同鎂原子一樣其所轉移的電子分散在整個α1硼烯中,因此與硼烯表面的作用范圍較大,而與鎂原子不同的是,B13在吸附過程中同樣也失去了0.06e的電子,因此B13與鈣原子存在一定的電子之間排斥力,這也是鈣原子吸附能小于鎂原子的另一個原因.三種堿土金屬的共同點在于,距離穩定位較遠的部分硼原子同樣也失去了較少的電子,并且α1硼烯在六邊形結構附近是極易得到電子的,且平面整體呈現離域性.
圖3為三種不同的堿土金屬吸附在α1硼烯最穩態上的一系列電子態密度(PDOS)圖,虛線代表費米能級.從(a)中可以看到主要是體系的s和p軌道都穿過了費米能級,說明吸附后的α1硼烯沒有帶隙,和純的硼烯一樣呈現金屬特性.從(b)、(c)、(d)圖中可以看到堿土金屬都是外層s軌道的電子轉移到了硼烯上,并與其產生了相互作用.鈹原子吸附在α1硼烯后的電子態密度產生了很大的變化,當鈹原子與硼原子相互作用時,其s軌道上的電子被部分激發,來到了原本空著的三個高能級p軌道,其s態和p態的電子同α1硼烯上硼原子上的電子相互作用產生了s-p以及p-p共價鍵. 鎂原子由于吸附在α1硼烯表面上的距離大于鈹原子,其s軌道上的電子只有一小部分被激發來到了p軌道同硼原子產生了共價鍵.鈣原子吸附在α1硼烯表面上的距離是最大的(2.532 ?),其原本占據在s軌道的電子絕大部分離開了原本的軌道了,來到了α1硼烯上,因此鈣原子與硼烯主要以離子鍵為主,這也解釋了為什么鈣的吸附能小于鎂.

圖3 堿土金屬在α1硼烯最穩態上的PDOS圖.(a)為α1硼烯以及三種堿土金屬吸附在α1硼烯上總的PDOS圖,(b)、(c)、(d)分別為孤立的鈹、鎂、鈣原子的PDOS圖以及吸附在α1硼烯上后鈹、鎂、鈣原子的PDOS圖Fig. 3 The PDOS diagrams of the alkaline earth metal in the most stable state of α1 borophene. (a) is the total PDOS diagram of α1 borophene and three alkaline earth metals adsorbed on α1 borophene, and (b),(c)and (d) are PDOS diagrams of free magnesium and calcium atoms and PDOS diagrams of beryllium, magnesium and calcium atoms adsorbed on α1 borophene, respectively.

圖4 鈹二聚體團簇吸附在α1硼烯上的前12種最穩構型及其與最穩定構型的相對能量差Fig.4 The first 12 most stable configurations of Be clusters adsorbed on α1 borophene and their relative energy differences with the most stable configuration
3.3堿土金屬雙原子團簇在α1硼烯表面的吸附性質
為了進一步考察多個堿土金屬原子吸附在α1硼烯上的幾何結構,穩定性以及對應的性質,我們同時對堿土金屬雙原子二聚體團簇在α1硼烯表面的吸附進行進一步的探索.顯然,二聚體團簇吸附硼烯的可能初始吸附位置要比單原子復雜的多,我們考慮了眾多可能的吸附位置和吸附方式,并進行了結構優化.找到了堿土金屬二聚體團簇吸附在α1硼烯上幾種穩定構型,并選取了吸附能最大的前十二種構型整理如圖4、5、6所示,排布順序按照吸附能由大到小排列.
如圖4所示當鈹團簇的初始試探位位于空的六邊形孔洞附近時,團簇首先會縮短鍵長,然后一個原子會慢慢來到六邊形空洞的中心,另一個來到B1或B5,這兩個都是穩定點,B5比B1相對更穩定.硼烯整體對鈹原子具有較強的吸引力,原子距離硼烯表面較近,其中一個鈹原子對B5或B1形成一定程度的擠壓,造成B5或B1略微下移.而當初始試探位在滿的六邊形孔洞周圍時,吸附過程中鈹團簇首先也會縮短鍵長,通過擠壓硼烯表面來達到平衡,但這種情況下,吸附能普遍不是很大.最后我們還研究了鈹團簇垂直于硼烯表面時的吸附情況,發現吸附能普遍不如前兩種,其原因推測距離硼烯較遠的那個鈹原子與硼烯基底幾乎沒有相互作用了,具體吸附情況如圖4所示.
圖5是鎂團簇吸附在α1型硼烯上的情況,我們發現吸附能最大的地方同樣也是其中一個鎂原子在Hollow位,而另一個鎂原子位于距離下一個Hollow位較近的地方或者其它吸附位(Top,Bridge,Triangle),最低能量構型中兩個鎂原子傾向于各自吸附于單個鎂原子的最穩定吸附位置(Hollow位).當初始試探位位于充滿的六邊形孔洞附近時,鎂團簇的吸附能就普遍不是很高.最后我們同樣也嘗試了垂直于硼烯表面的情況,發現吸附能是最低的,這些與鈹團簇的吸附情況是大致一樣的.而與鈹團簇不同的是,鎂團簇的亞穩態點明顯多于鈹團簇,在很多位置都能穩定(如圖5所示).此外,鎂團簇吸附在α1硼烯上時,最穩定的幾種吸附構型大多傾向于一個鎂原子吸附在Hollow位,即使改變初始構型的吸附點,優化后一個鎂原子始終穩定在Hollow位,從中看出α1硼烯的Hollow位對單個鎂原子具有很強的吸引作用,這個和前面單個鎂原子在Hollow位具有較大的吸附能(21.321 eV)相吻合.

圖5 鎂二聚體團簇吸附在α1硼烯上的前12種穩定構型及其與最穩定構型的相對能量差Fig.5 The first 12 most stable configurations of Mg clusters adsorbed on α1 borophene and their relative energy differences with the most stable configuration
圖6為鈣團簇的吸附情況,發現當一個鈣原子吸附在靠近B13附近,另一個鈣原子吸附在六邊形孔洞中心的附近時,體系的吸附能是最大的,這分別是單個鈣原子吸附時,吸附能最大的兩個位置.同鎂團簇吸附情況相似的是,當兩個鈣原子都十分靠近六邊形孔洞中心的附近時,體系的吸附能相對是較大的,當鈣團簇吸附在被充滿的六邊形孔洞附近時吸附能次之,垂直情況同其它堿土金屬一樣是最小的.

圖6 鈣二聚體團簇吸附在α1硼烯上的前12種最簡構型及其與最穩定構型的相對能量差Fig. 6 The first 12 most stable configurations of Ca clusters adsorbed on α1 borophene and their relative energy differences with the most stable configuration
從表2以及圖7中我們不難發現堿土金屬雙原子團簇吸附在α1硼烯上后,團簇之間的距離會縮短,二聚體團簇的吸附能是稍大于單個堿土金屬的吸附能的,然而除了鈹原子,團簇的平均吸附能是明顯小于單個原子的吸附能,可見α1硼烯更傾向于單個原子的吸附.因此我們還計算了兩個堿土金屬各自吸附在最穩態時的情況(單個鈣原子吸附時Near B13和Near Hollow兩個位置的吸附能相差很小,兩個堿土金屬原子吸附后原子位于Near Hollow的吸附能稍大于Near B13),研究發現當兩個堿土金屬原子孤立吸附在硼烯后,其吸附能相比于二聚體團簇反而稍大一些,由此我們推測當堿土金屬原子吸附在α1硼烯上時,分散的單原子吸附在能量上是較有利的,吸附原子之間不會傾向于聚集成團簇.
4結 論
本文采用基于密度泛函理論的Materials Studio計算軟件中的CASTEP模塊對堿土金屬吸附α1硼烯的最低能量吸附位置、結構穩定性和相互作用進行了系統的計算和研究. 首先對堿土金屬(鈹、鎂和鈣)單原子、二聚體團簇、雙原子吸附在α1硼烯上最低能量構型進行了鑒定和討論,在此基礎上研究了堿土金屬原子與硼烯的相互作用.結果表明:(1)單個鈹原子和鎂原子吸附在α1硼烯Hollow位是體系吸附能最大的情況,而鈣原子在B13附近時體系的吸附能最大.(2)相對于鈹原子情況,鎂、鈣原子吸附在α1硼烯上的吸附能較大,說明α1硼烯在儲鎂和儲鈣方面存在應用潛力.(3)通過電荷轉移和態密度等性質分析,發現鈹原子吸附在最穩態時可以與α1硼烯產生s-p以及p-p共價鍵,鈹與硼原子的共價鍵最明顯.鎂原子是離子鍵和共價鍵的混合成鍵方式,而鈣原子則主要是離子鍵形式.(4)對于二聚體吸附情況,雙原子鈹團簇的最穩態為一個鈹原子在Hollow位,另一個在B1附近,鎂團簇的最穩態為一個在Hollow位,另一個鎂原子在其它的Hollow位附近,鈣團簇的最穩態為一個在B13附近,另一個在Hollow的中心附近.(5)二聚體團簇吸附能大于單原子吸附能但小于雙原子吸附情況,表明堿土金屬原子吸附在α1硼烯上時,吸附原子之間不會傾向于聚集成團簇,在穩定性上反而更傾向于分散的單原子吸附方式.研究結果和前人堿金屬吸附的情況進行了對比和討論,這些研究結果豐富了二維硼烯的體系和結構,并為開發以二維硼烯為基礎的功能性納米材料提供參考價值.

表2 堿土金屬雙原子團簇吸附在α1硼烯的吸附能,Lc為真空中孤立的雙原子團簇間的距離,D為實際優化后兩個原子間的距離,Et為總的能量,Eb1為二聚體團簇的吸附能,Eb2為兩個堿土金屬原子分別吸附在最穩態的吸附能
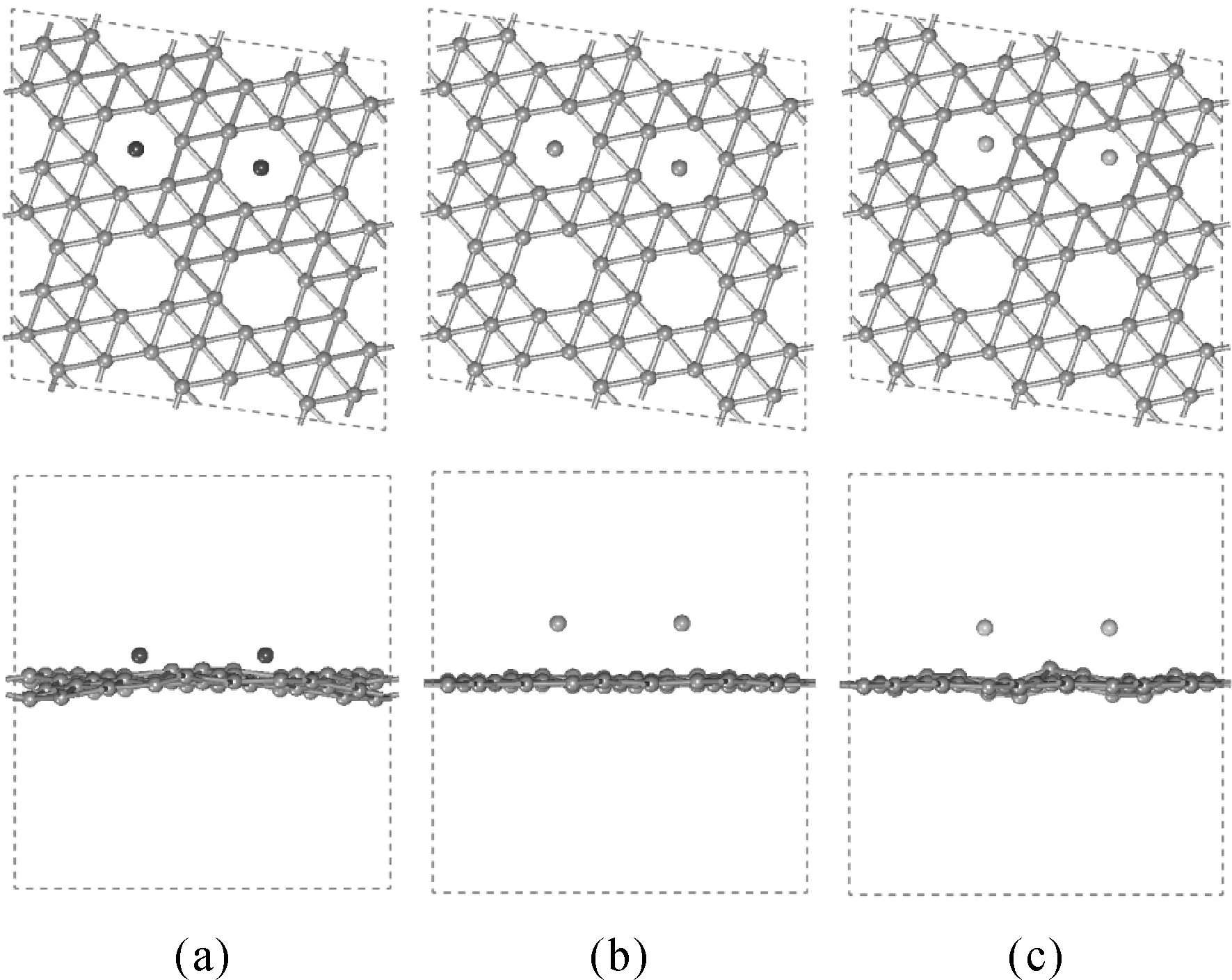
圖7 (a)、(b)、(c)分別為兩個鈹、鎂、鈣原子各自吸附在最穩態上的俯視圖和側視圖Fig. 7 (a), (b), and (c) are top and side views of the two beryllium, magnesium, and calcium atoms adsorbed on the borophene in the most stable system, respectively
