基于第一性原理計算H2S、CO2、CH4在Fe-MoS2上的吸附性能
2020-05-13 08:45:14劉建儀陳奕兆
原子與分子物理學報 2020年4期
關鍵詞:結構
劉建儀, 陳奕兆, 謝 泱, 袁 華, 劉 淼
(1. 西南石油大學 油氣藏地質及開發工程國家重點實驗室, 成都 610500; 2. 西南石油大學 石油與天然氣工程學院, 成都 610500)
1引 言
世界上的高含硫氣藏的儲量不可忽略,尤其是在常規能源開發技術日漸完善下,安全高效的開發此類氣藏具有很高的經濟價值,同時回收的硫元素也可用于化學工業. 由于施工成本等各種因素,難以清潔井場中的含硫天然氣,需要通過管道將其運輸到遠程凈化設備中清潔. 在這個過程中,井場和輸送管道將不可避免地溢出H2S和CO2,這極大地危害了周圍環境的安全并腐蝕管道[1-3]. 因此,必須找到一種有效的方法來去除溢出的H2S,CO2. 目前,文獻研究了Mo(110),TiC的吸附性能,以及α-Fe2O3(001)和TiO2對H2S的吸附和離解性能[4-7]. Leonardo Hadlich de Oliveira報道了NAY沸石上的H2S吸附[8]. Noushin Osouleddini發現改性石墨烯表面含有TCNE分子作為CO2和CH4的良好吸附劑[9]. 近年來,由于石墨烯等二維層狀納米材料的興起,二硫化鉬(MoS2)由于其優異的機械、電子和光學性質而受到許多研究人員的廣泛關注[10-12],并且MoS2單層具有對許多氣體敏感的特征,諸多學者在這方面進行了大量研究[11,13,14]. 趙世軍和薛建明發現NO,NO2,SO2與MoS2的結合被認為是所有被考慮的氣體分子中吸附能力最強[13]. 大量研究證實,與MoS2單層相比,摻雜的MoS2單層結構更能改善對氣體分子的吸附性能[15,16],其中部分研究表明摻雜Fe,Pt,Si,Ni等原子顯著改善MOS2的吸附性能[11,17-22]. 通過文獻的調查,MoS2中Fe的吸附式摻雜結構研究較少,同時相關研究中對高硫氣藏特有的氣體(CO2和H2S)吸附計算尚未見有報道. 本文采取第一性原理計算了H2S,CO2,CH4氣體在Fe-MoS2上的吸附能,電荷轉移,電子密度差等相關參數,得到了較好的結果.
2計算參數的設置
所有的計算都是基于MS軟件的DOML3模塊[23]. 建立了4×4×1的超晶胞MoS2,為防止周期性計算引起超晶胞層之間的相互作用,在C方向加入10 ?真空層.在執行結構優化時,使用Perdew-Burke-Ernzerhof(PBE)的廣義梯度近似(GGA)來處理電子之間的相互作用能[24],并且使用單個有效勢替代內核電子(DFT semi-core Pseudopots),在內核處理中引入相對論效應. 采用雙數值軌道基組和軌道極化函數(DNP)的基組設置,DNP數值設為4.4. 能量收斂精度設置為2×10-5/?,內部原子間相互作用力不超過4×10-3Ha/?,最大位移設為5×10-3?,自洽場(SCF)收斂精度為1×10-5Ha. 在所有計算中都考慮了自旋極化效應. 布里淵區采用Monkhorst-Pack網格,K點為5×5×1[25].
Morphology模塊計算表明MoS2最易暴露的面為(003),因此選擇(003)面為研究對象. 在文章中計算參數定義如下:
氣體分子在Fe-MoS2上的吸附能為:
Eads=EFe-MoS2/gas-EFe-MoS2-Egas
(1)
電荷轉移表示為:
Qt=Qa-Qb
(2)
3結果與討論
3.1H2S、CH4、CO2、MoS2、Fe-MoS2的結構
圖1的(a)-(c)表明了H2S、CH4、CO2的分子結構. 基于Mulliken 方法,H2S分子中H原子帶有0.073e的正電荷,S原子帶有-0.174e的負電荷;CH4分子中H原子帶有0.055e的正電荷,C原子帶有-0.218e的負電荷;CO2分子中O原子帶有-0.251e的負電荷,C原子帶有0.502e的正電荷. (d)圖表明了MoS2的結構.Mo-S鍵長為2.418 ?.
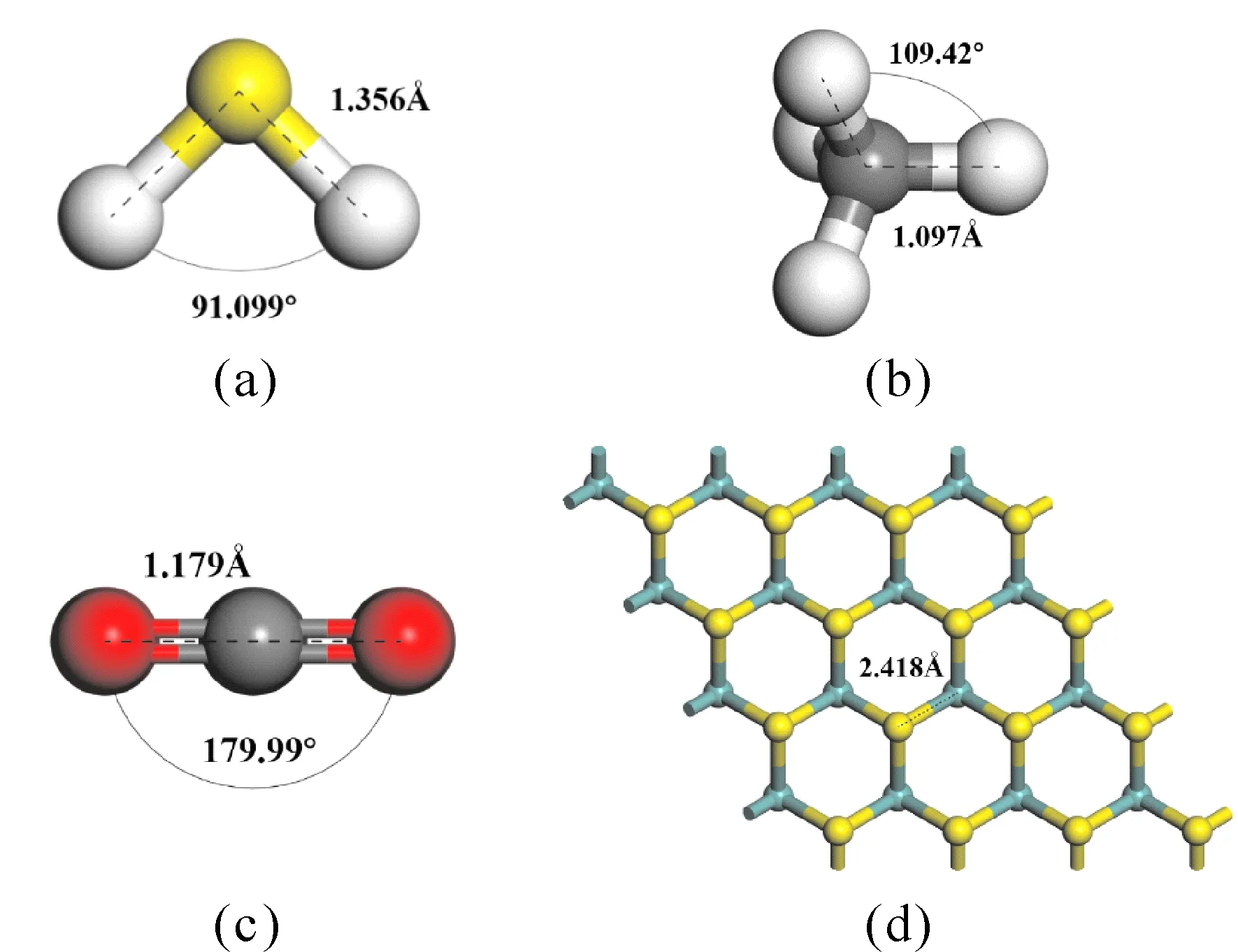
圖1 H2S、CH4、CO2、MoS2的結構Fig. 1 The Structure of the H2S, CH4, CO2 and MoS2
考慮到MoS2材料的對稱性,對于Fe吸附式摻雜在MoS2上存在三種可能的位置. Mo原子的頂部、在S原子的頂部、在MoS2六圓環的中心上方. 圖2表示了這些位置,表1給出了吸附式摻雜后的相關吸附能. 計算結果表明Fe原子摻雜在MoS2中最穩定的位置是在Top-Mo,吸附能為-3.804eV.

圖2 Fe摻雜在MoS2上的三種結構Fig. 2 Three structures of Fe doped on MoS2
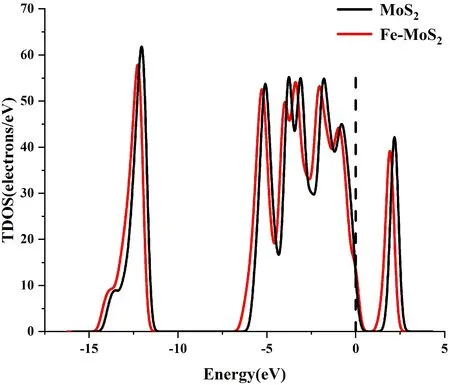
計算材料的總態密度(TDOS)和分波態密度(PDOS),定義費米能級EF=0 eV,進一步分析Fe原子摻雜在MoS2的摻雜機理. 圖3所示,摻雜Fe原子后,材料的TDOS整體呈左移趨勢,表明摻雜原子前后系統總能量發生了改變. 通過對PDOS波形分析,Mo原子的4d軌道與Fe原子的3d軌道在-5.5eV, -5eV, -3eV, -0.5 eV發生了重疊,表明Fe原子與Mo原子之間較強的軌道雜化作用.
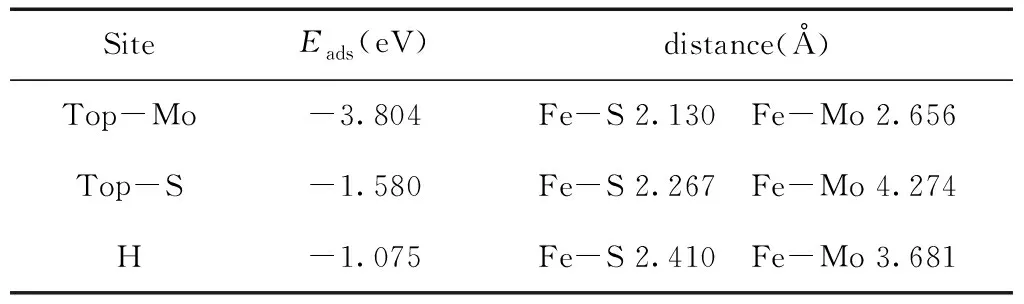
表1 不同摻雜位置的吸附能及距離
(a)
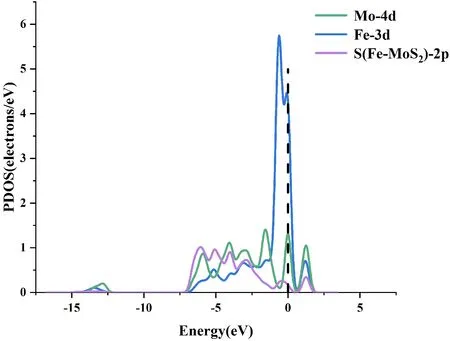
(b)圖3 (a)摻雜前后體系TDOS; (b)Mo、Fe、S原子的PDOSFig. 3 (a) The TDOS of system doped before and after;(b)The PDOS of Mo, Fe and S
與此同時,也分別計算了Fe-MoS2和MoS2的帶隙.帶隙的減小,表明電子由價帶激發到導帶更加容易,體系更加穩定.

表2 摻雜前后材料的帶隙
3.2H2S在Fe-MoS2上的吸附
進行了三次計算以驗證H2S在Fe-MoS2上的吸附.M1,M3是H2S平行于Fe-MoS2,M2則是H2S垂直于Fe-MoS2. 這三種狀態下的吸附參數都趨于一致. 表3給出了相關的參數信息,最高的吸附能量是M3系統,吸附能量為-1.512eV,吸附后H2S分子與Fe原子的距離為2.260?. 分析氣體分子吸附前后的總電荷數,氣體分子的正電荷在吸附后增多,計算了Fe原子在吸附前后的電荷數,Fe原子電子增加. 表明在吸附過程中,電子由氣體分子轉移到Fe原子附近.
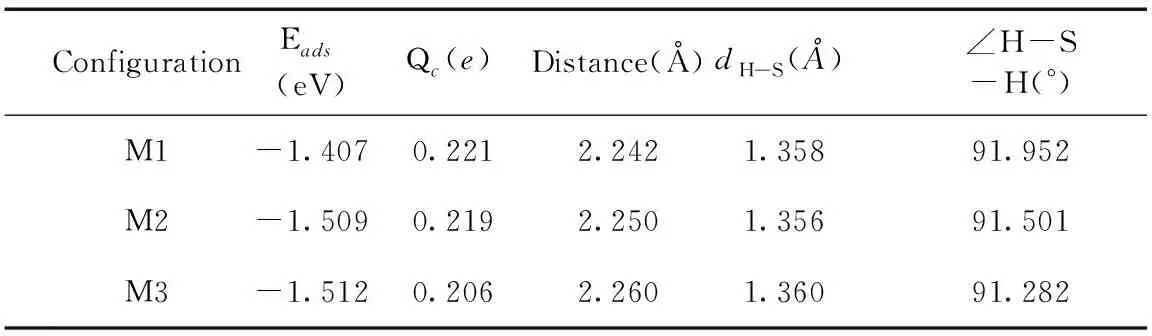
表3 H2S在Fe-MoS2上吸附的相關參數

表4 吸附前后Fe原子和H2S分子電荷數
圖4是優化后H2S分子吸附在Fe-MoS2的M3結構.吸附后,MoS2單層與H2S分子均發生了細微變化. 對于MoS2單層結構而言,Fe-Mo鍵長距離增大了0.066 ?,Fe-S(MoS2)的鍵長增大了0.023 ?,鍵長的增大表明了H2S的吸附使得Fe與MoS2的鍵合能力變弱. 對于H2S分子,鍵長略微增大0.002 ?,鍵角也較吸附前略微增加0.18°,表明吸附對氣體分子結構影響很小.
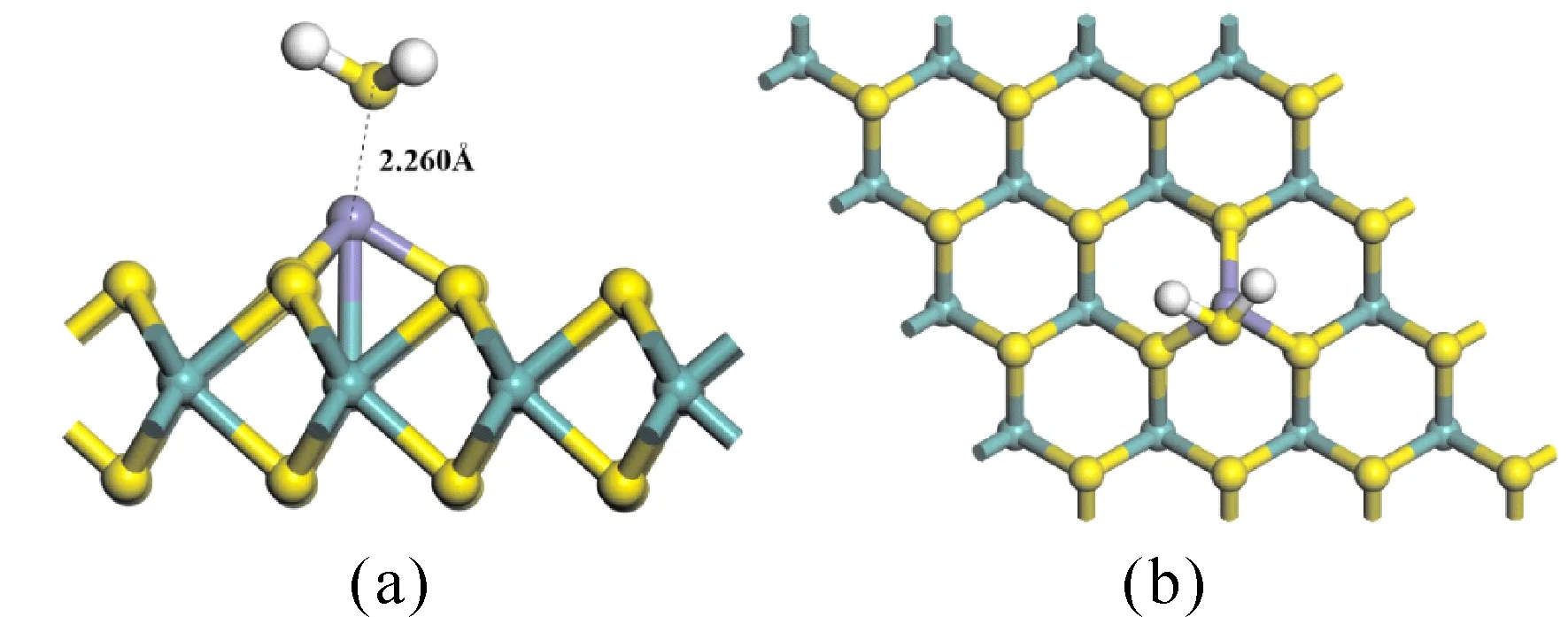
圖4 優化后H2S吸附在Fe-MoS2上的側視圖與俯視圖Fig. 4 The side view and top view of optimized H2S adsorption on Fe-MoS2
圖5是對H2S吸附在Fe-MoS2上的態密度圖. 從TDOS圖上顯示,波形有輕微的左移,且在波峰處有所變化,表明吸附后材料發生了能量的變化. 對于PDOS, S原子的2p軌道與Fe原子的3d軌道在-4eV, -6eV, -7.5eV等位置有明顯的波峰重疊,表明S原子與Fe原子發生了軌道雜化作用.
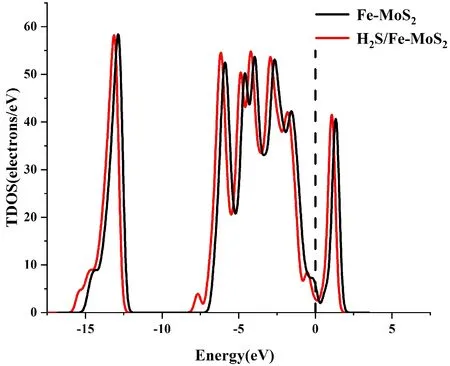
(a)
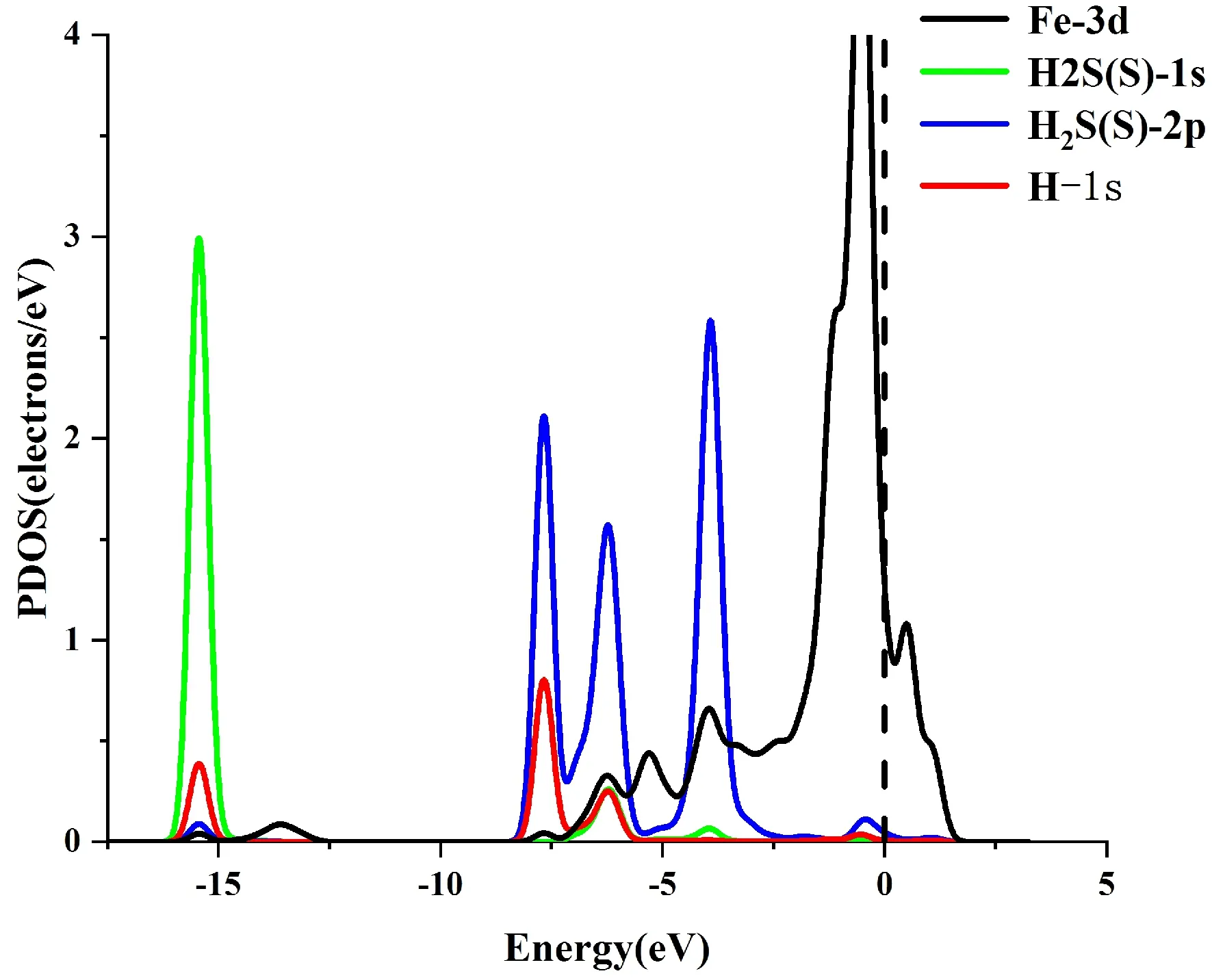
(b)圖5 (a)吸附H2S前后的TDOS; (b)吸附H2S后的PDOSFig. 5 (a)The TDOS of system before and after adsorpting of H2S; (b)The PDOS of system after adsorpting of H2S
為了進一步分析H2S與Fe-MoS2之間的電荷轉移機制,圖6給出了Fe-MoS2吸附H2S的差分電荷密度圖,H2S失去電子給Fe-MoS2,其中Fe原子的存在促進了H2S失去電子的能力.
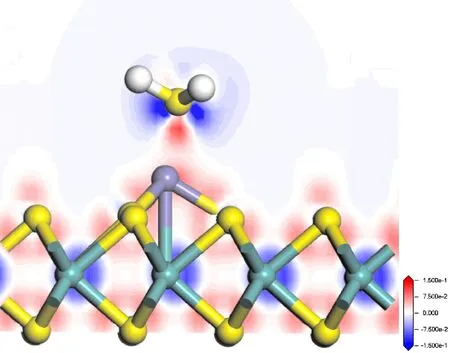
圖6 H2S吸附在Fe-MoS2上的差分電荷密度Fig. 6 The differential charge density of H2S adsorbed on Fe-MoS2
3.3CH4在Fe-MoS2上的吸附
進行了三次計算以驗證CH4在Fe摻雜在MoS2上的吸附,M1,M3是H原子垂直于Fe原子,M2是C原子垂直于Fe原子. 表5給出了相關的參數信息.最高的吸附能量是M1系統,吸附能量為-0.688eV.吸附能和電子轉移都遠遠小于H2S在Fe-MoS2上的吸附參數. 表明這種吸附是一種較弱的作用力,屬于物理吸附范疇.
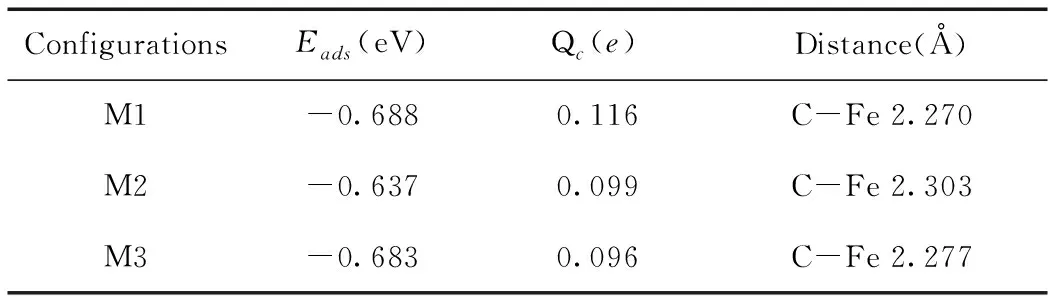
表5 CH4吸附與Fe-MoS2上的相關吸附參數
圖7是優化完成的CH4吸附在Fe-MoS2的結構圖. 吸附后CH4中C原子與Fe原子距離為2.270 ?. CH4分子的結構沒有明顯的變化,靠近Fe原子一側的C-H鍵長略微增加了0.024 ?,另一側的C-H鍵長保持不變. Fe原子與Mo原子之間的鍵長增加了0.075 ?,Fe原子與相鄰S原子的鍵長增加了0.028 ?.這些略微的變化都表明這種吸附作用力十分微弱.
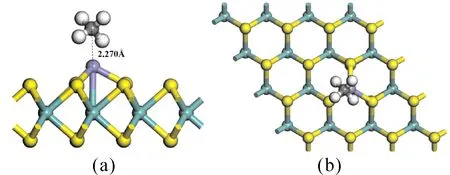
圖7 優化后CH4吸附在Fe-MoS2上的側視圖與俯視圖Fig. 7 The side view and top view of optimized CH4 adsorption on Fe-MoS2
圖8給出了吸附后的TDOS與PDOS. 從TDOS中,吸附CH4前后體系的TDOS基本重合,表明吸附對體系沒有產生能量的變化.從PDOS中,Fe原子與C原子和H原子重疊的部分很小,表明這種吸附不存在強力的相互作用,這與前面的數據分析結果相吻合.
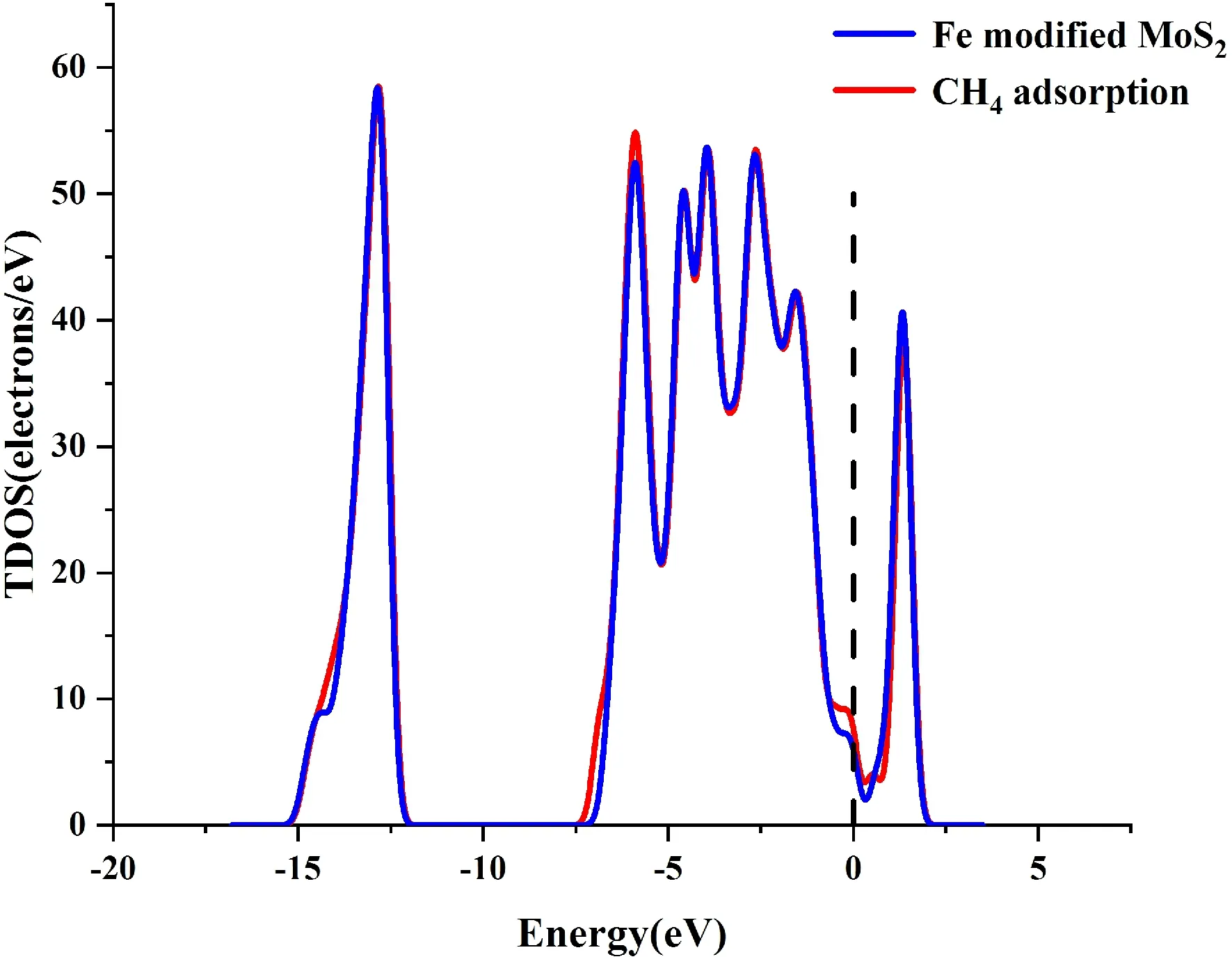
(a)
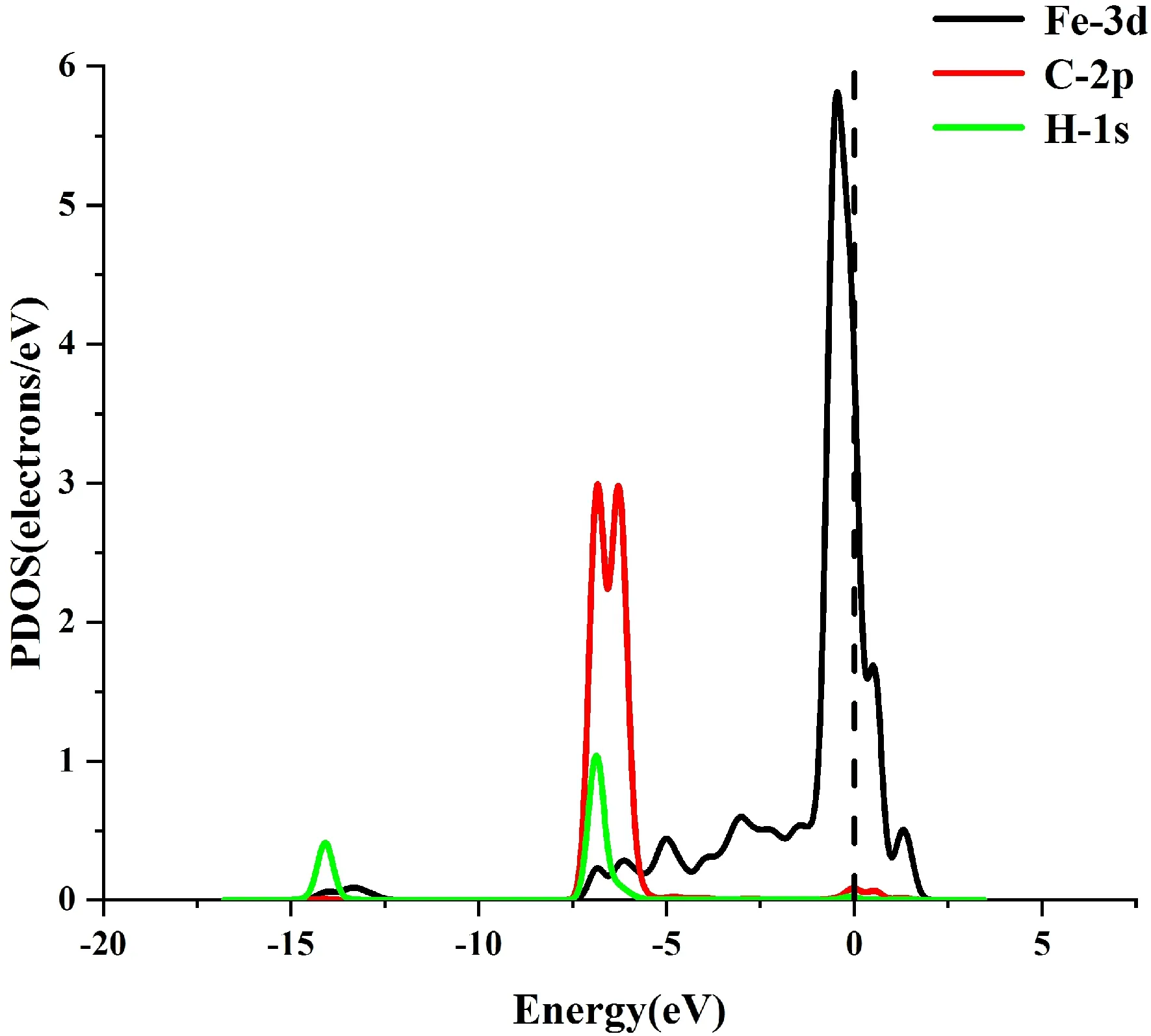
(b)圖8 (a)吸附CH4前后的TDOS;(b)吸附CH4后的PDOSFig. 8 (a)The TDOS of system before and after adsorpting of CH4; (b)The PDOS of system after adsorpting of CH4
為了進一步分析CH4與Fe-MoS2之間的電荷轉移機制,圖9給出了Fe-MoS2吸附CH4的差分電荷密度,可以發現電子轉移較弱. 表明CH4與Fe-MoS2之間相互作用力很弱,屬于物理作用力.

圖9 CH4吸附在Fe-MoS2上的差分電荷密度圖Fig. 9 The differential charge density of CH4 adsorbed on Fe-MoS2
考慮到CO2分子的對稱性,在對CO2進行吸附計算時,在M1,M2系統中,CO2分子的O原子垂直于Fe原子上方,Fe原子的取向為O原子.M3,M4系統則是水平放置于Fe原子上方.
相關吸附參數已在表6中給出,最高的吸附能為-2.416 eV,較M1,M2的結果相對比,M3,M4的結果表明CO2在Fe-MoS2的吸附可能存在一種更加穩定,作用力更強的吸附. 從電荷轉移也可以證實這點.
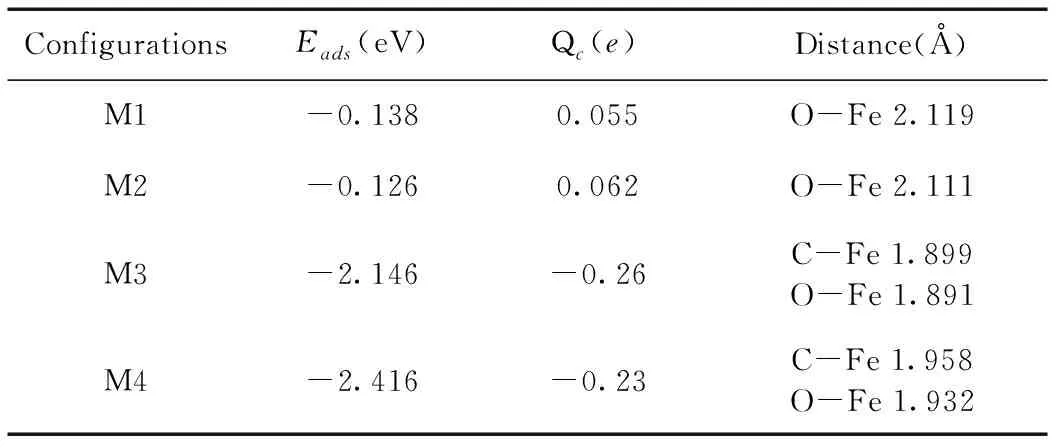
表6 CO2吸附在Fe-MoS2上的相關吸附參數
圖10給出了M4吸附結構,CO2由直線型分子變化為V型分子,鍵角由179.99°減小到 143.654°,這表明著在C原子附近產生了大量的電子,電子的排斥作用使得鍵角發生了改變. Fe原子與O原子和C原子的距離分別為1.932 ?和1.958 ?. Fe原子與Mo原子的距離增加了0.156 ?,Fe原子與S原子的距離增加了0.057 ?,這些鍵長的變化表明了吸附作用比CH4與H2S分子的影響更加強烈. 從電子轉移的角度來看,在M3與M4系統中,CO2是電子的受體,Fe原子是電子的給體,這與M1,M2系統恰好相反.
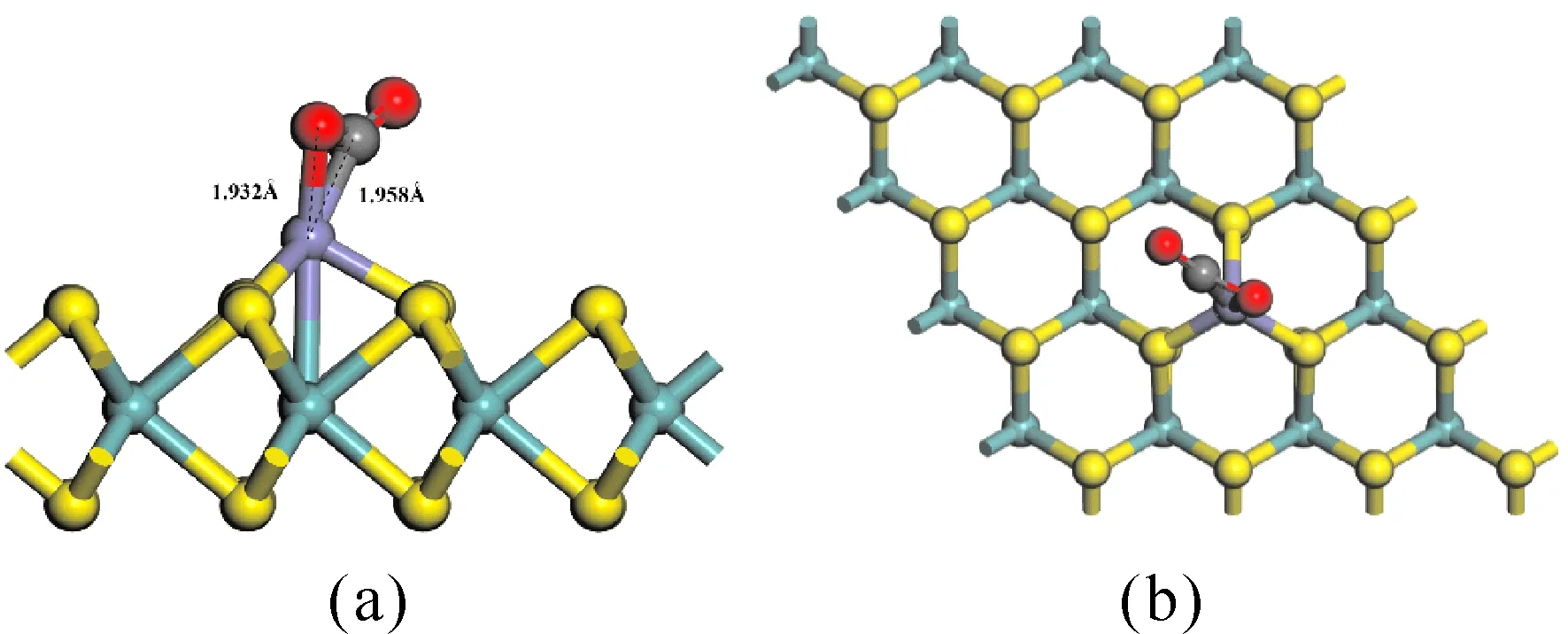
圖10 CO2吸附在Fe-MoS2上的吸附結構側視圖與俯視圖Fig. 10 The side view and top view of optimized CO2 adsorption on Fe-MoS2
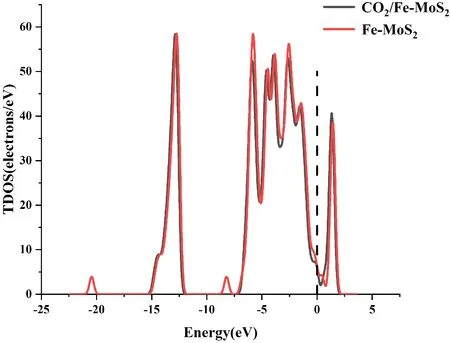
圖11給出了這種吸附結構的TDOS與PDOS.對于TDOS,在-8eV,-20.5 eV處發生了突變,這主要是O原子與C原子的2p軌道的貢獻,表明了體系能量發生了變化.
(a)
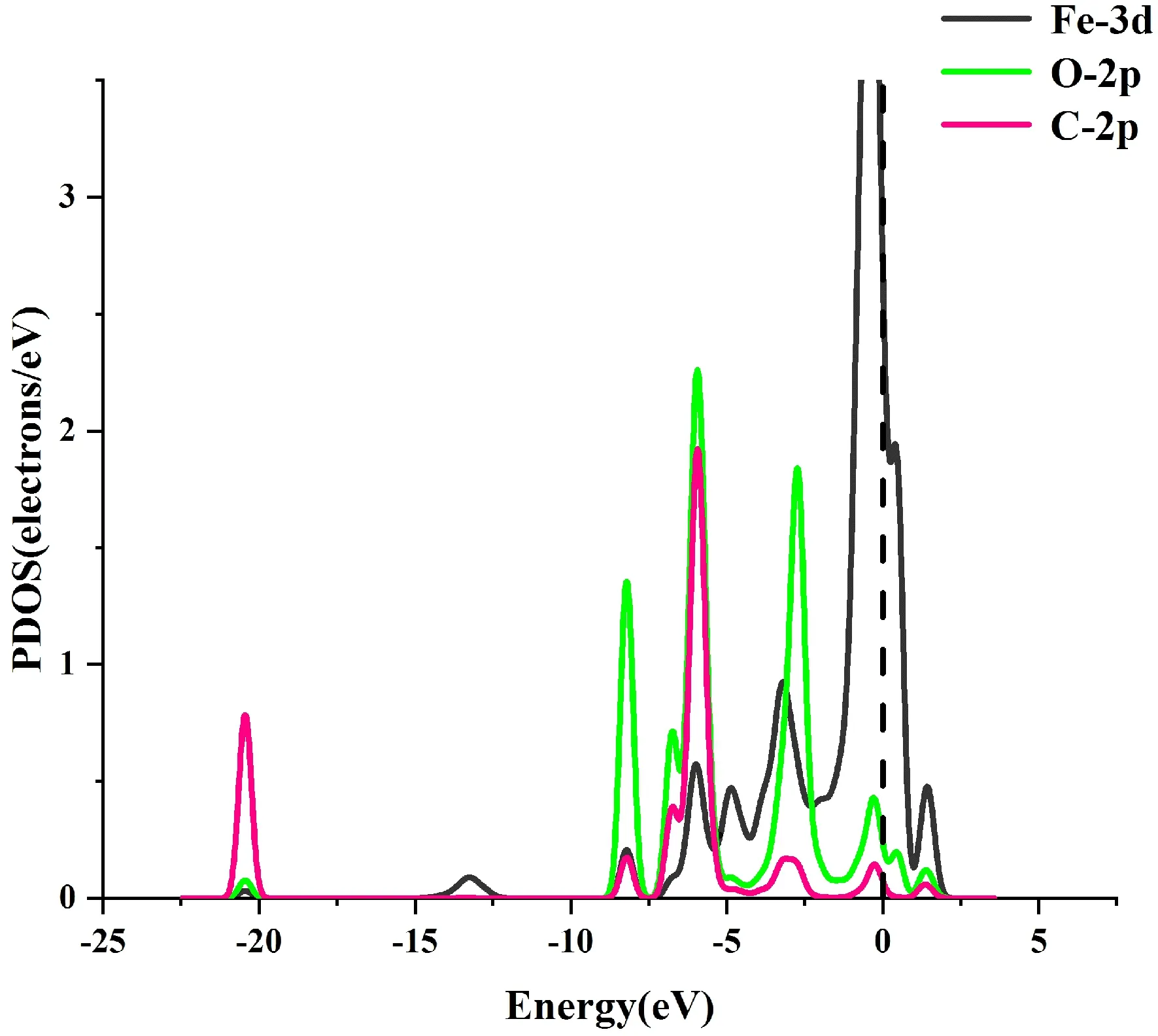
(b)圖11 (a)吸附CO2前后的TDOS;(b)吸附CO2后的PDOSFig. 11 (a)The TDOS of system before and after adsorpting of CO2;(b)The PDOS of system after adsorpting of CO2
對于PDOS,在-4.5 eV,-5.5eV,-3eV,-2.5eV,-0.5 eV等能量處有峰的重疊,表明了Fe原子與C原子O原子之間存在軌道雜化作用.
圖12給出了Fe-MoS2吸附CO2后的差分電荷密度圖,CO2得到電子,Fe原子附近電子密度減小.這與前面的電子轉移分析結果相吻合.
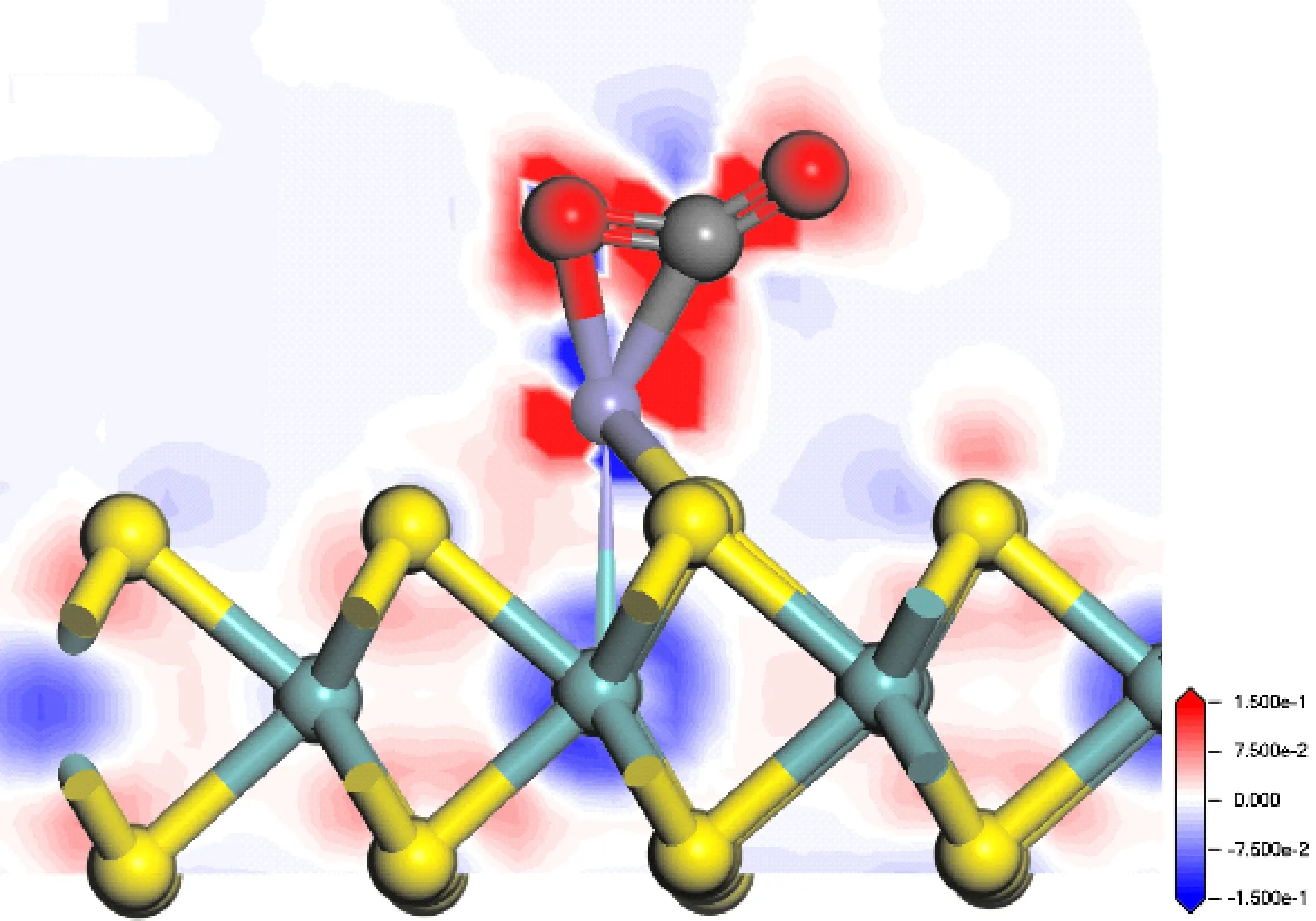
圖12 CO2吸附在Fe-MoS2上的差分電荷密度圖Fig. 12 The differential charge density of CO2 adsorbed on Fe-MoS2
4結 論
(1)計算了Fe原子在單層MoS2上存在的三種可能吸附的位點,得到了最穩定的吸附位置,位于Mo原子的頂部.
(2)分別計算了H2S,CH4,CO2三種氣體在Fe-MoS2上的吸附結構,得到了相應的吸附參數.H2S,CH4,CO2在Fe-MoS2上的吸附能分別為-1.512eV,-0.688 eV,-2.416 eV.
(3)H2S和CO2在Fe-MoS2上是一種化學吸附,CH4在Fe-MoS2上是一種物理吸附.對于CO2在Fe-MoS2上的吸附,計算得到了一種較強的吸附結構.
(4)Fe原子摻雜MoS2的材料對三種氣體有著不同的吸附特性,表明這種材料有可能被開發成為一種氣體報警裝置或是一種良好的氣體吸附劑.
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
