鋅在林格氏液中的體外長期腐蝕降解行為
2020-05-13 01:55:46唐帥張文泰錢軍余鮮鵬莫小山黃楠萬國江
無機材料學報 2020年4期
唐帥, 張文泰, 錢軍余, 鮮鵬, 莫小山, 黃楠, 萬國江
鋅在林格氏液中的體外長期腐蝕降解行為
唐帥, 張文泰, 錢軍余, 鮮鵬, 莫小山, 黃楠, 萬國江
(西南交通大學 材料科學與工程學院, 材料先進技術教育部重點實驗室, 成都 610031)
鋅基可降解生物材料與已被廣泛研究的生物可降解材料(鎂和鐵)相比, 具有更合適的生物降解速率, 因而近年來受到了廣泛的研究和關注。然而, 鋅在模擬體液中的長期腐蝕降解行為尚不明確。本研究采用電化學腐蝕測試、表面化學成分分析及降解模式演變觀察相結合的方法, 系統研究了鋅在林格氏液中浸泡56 d的腐蝕演化過程。根據電化學結果顯示, 鋅的腐蝕速率i在浸泡過程中基本保持穩定, 約為0.06~0.10 mm/a; 失重法測定腐蝕速率為0.3 mm/a到0.5 mm/a。浸泡過程中生成的腐蝕產物主要為Zn5(CO3)2(OH)6和CaCO3, 為較致密的細條花棒狀和塊狀產物層, 且隨著浸泡時間延長逐漸累積。去除腐蝕產物后發現, 樣品表面出現較嚴重的局部腐蝕, 且腐蝕溝槽的尺寸隨浸泡時間的延長而增大, 浸泡42 d腐蝕溝槽寬約為10 μm。本研究為鋅基可降解生物材料后期表面改性及潛在生物醫學應用提供了數據積累和研究基礎。
可降解鋅; 長期腐蝕行為; 電化學測試; 表面化學
近年來, 鋅基金屬與其它可降解金屬相比, 具有更合適的生物降解速率的優勢, 正成為最具潛力的生物可降解植入材料[1-2]。鋅是人體內第二位必需微量元素, 成人和嬰兒的鋅推薦日攝入量分別為8~11 mg/d和2~3 mg/d[3], 因此鋅植入物在體內降解過程中產生的鋅離子安全濃度窗口大, 不易產生系統毒性, 可適應不同服役部位和生理環境。同時, 鋅參與了眾多生理代謝過程, 具有一定的生理功能[4-6], 包括信號傳導、細胞凋亡調節、抗動脈粥樣硬化和促成骨礦化以及骨細胞生長和愈合等。當前針對鋅基材料力學性能不足的合金化工作已取得了諸多進展, 大量合金體系已可以滿足臨床力學性能要求[7-9]。鋅(?0.762 V(SHE))的標準電極電位介于鎂(?2.34 V(SHE))和鐵(?0.44 V(SHE))之間, 相對于鎂在體內的降解速率過快和鐵的降解速率過慢, 鋅在理論上具有更適宜的生物降解速率。
一般來說, 生物可降解植入體需在人體內停留3~6個月甚至更長時間[10], 深入了解其在植入部位服役過程中的降解行為十分重要, 而利用體外模擬生理環境預測鋅在體內的長期腐蝕行為是一種行之有效的方法, 同時也對進一步的表面改性提供理論基礎。陳英奇等[11]對比了鎂、鋅和鐵在磷酸鹽緩沖液中的降解行為, 發現暫態測試中鋅的開路電位和降解速率位于鎂和鐵之間, 而浸泡過程中, 鋅的降解速率比鎂和鐵快, 這與理論不一致。而Karin等[12]研究了純鋅在鹽溶液、血漿和全血中3 d的降解行為, 發現血漿和全血中樣品均勻腐蝕的表面上主要是由無機鹽和有機分子組成的腐蝕產物; 磷酸鹽緩沖液中樣品為局部腐蝕, 表面主要由磷酸鋅組成的厚而多孔的腐蝕產物層; 而林格氏液中, 樣品表面淺坑界面上形成了碳酸鋅凝膠狀產物層, 更接近于血漿和全血中的降解行為, 故對于鋅的體外研究來說, 林格氏液要優于磷酸鹽緩沖液。近期越來越多的研究開始關注純鋅在模擬體液中的腐蝕行為[13-15], 然而大多數只局限于短期(<14 d)腐蝕降解行為, 缺乏長期腐蝕降解過程中電化學腐蝕、腐蝕產物演變、腐蝕模式和腐蝕機制等方面的系統研究。
基于此, 本研究采用長期浸泡(56 d)的方式, 研究鋅在模擬體液(林格氏液)中的腐蝕演化行為, 包括電化學腐蝕演化規律、表面腐蝕產物分析和腐蝕產物去除后的腐蝕模式演化。
1 實驗方法
1.1 樣品制備
將純鋅棒(99.99%)切成直徑10 mm, 厚度1.8 mm的圓片, 依次用600#、1000#、1500#和2000#的SiC砂紙研磨純鋅表面, 用酒精和去離子水超聲清洗打磨后的鋅片(每次5 min, 共3次), 最后用洗耳球吹干置于真空干燥箱中備用。本研究中電解質溶液為林格氏液(9 g/L NaCl, 0.43 g/L KCl, 0.2 g/L NaHCO3, 0.24 g/L CaCl2), 所有試劑均為分析純。林格氏液的pH為7.4 ± 0.2。
1.2 電化學腐蝕表征
采用IM6電化學工作站測量樣品動電位極化曲線(Potentiodynamic polarization curves, PDP)和電化學阻抗譜(Electrochemical Impedance Spectroscopy, EIS)表征電化學腐蝕。電化學測試采用三電極系統裝置, 包括飽和甘汞參比電極(Saturated Calomel Electrode, SCE), 鉑對電極(Platinum sheet with dimensions of 1.5 cm ×1.5 cm)和工作電極(Zn samples)。工作電極樣品背面通過銅導線連接電化學工作站, 并用硅橡膠密封, 暴露待測試表面面積為0.79 cm2。在PDP和EIS測試前, 樣品在林格氏液中浸泡至少30 min, 以確保其表面處于穩定狀態, 然后置于(37±0.5) ℃水浴鍋中進行測試。對于動電位極化測試, 電位掃描區間為開路電位正負0.5 V, 掃描速度為1 mV/s。采用塔菲爾外推法對極化曲線進行計算, 得到樣品的腐蝕電位corr、腐蝕電流密度corr和陰極塔菲爾斜率c。根據標準ASTM G102-89[16], 腐蝕速率(i)計算公式如下:
i=14.93corr(1)
其中i、corr分別表示腐蝕速率(mm/a)和腐蝕電 流密度(mA/cm2)。電化學阻抗譜測試頻率范圍為100 kHz到0.01 Hz, 測試所得數據用軟件進行擬合分析。每組樣品重復4次電化學測試以符合統計學要求。
1.3 浸泡降解實驗
將打磨后的樣品浸泡在(37±0.5) ℃的林格氏液中, 在浸泡7、14、28、42和56 d時依次取出部分樣品, 研究其長期降解演化行為。每4個平行樣品設為一組, 浸泡在400 mL的林格氏液中, 每3 d更換一次溶液。浸泡結束后取出樣品, 用去離子水清洗三次并置于真空干燥箱中備用。用上述方法測試樣品的電化學腐蝕行為。采用體式顯微鏡(Stemi 2000-C, ZEISS, Germany)和場發射掃描電子顯微鏡(SEM, Quanta FEG 250, FEI, USA) 表征樣品形貌。通過X射線衍射(XRD)和X射線光電子能譜(XPS)分析表面腐蝕產物的結構和成分。XRD測試采用CuKα射線源, 2記錄范圍為10°~90°, 步長0.033°。XPS測試采用Al Kα (1486.6 eV) X射線源。根據標準ASTM G1-03, 將樣品浸泡在200 mg/mL CrO3溶液中(80 ℃) 1 min去除表面腐蝕產物, 并通過SEM觀察去除腐蝕產物后的表面形貌。將去除腐蝕產物前后的樣品浸泡在液氮中0.5 h后, 脆斷制作截面, 采用SEM和EDS線掃描分析截面的形貌和腐蝕產物的元素含量。對樣品浸泡前與去除腐蝕產物后進行稱重, 并根據標準ASTM G31-72計算腐蝕速率[17], 公式如下:
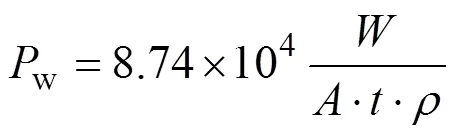
其中,w、、、、分別表示腐蝕速率(mm/a)、失重(g)、暴露面積(cm2)、浸泡時間(h)和密度(g/cm3)。
2 結果與討論
2.1 電化學腐蝕行為與分析
鋅在林格氏液中浸泡不同時間后的動電位極化(PDP)曲線如圖1所示。鋅的自腐蝕電位在浸泡前 約為?1.06 V(vs SCE), 浸泡過程中在?0.99 V(vs SCE)到?1.06 V(vs SCE)范圍內波動。對于活性金屬, 通過陰極區極化曲線計算塔菲爾斜率更為可靠[18]。計算結果顯示, 鋅浸泡后的自腐蝕電流密度均比浸泡前的電流密度小。在浸泡14 d后, 腐蝕電流密度從42.7 μA?cm–2逐步減小到6.2 μA?cm–2, 并在浸泡28 d后達到最低值4.9 μA?cm–2, 在浸泡42和56 d時保持穩定在5.7 μA?cm–2(圖2(a))。圖2(b)為根據自腐蝕電流密度計算得出的腐蝕速率i, 相比于浸泡前, 鋅浸泡后的腐蝕速率明顯降低, 從0.64 mm/a下降到0.08 mm/y, 并在整個浸泡過程中保持穩定, 約為0.06 mm/a到0.1 mm/a。
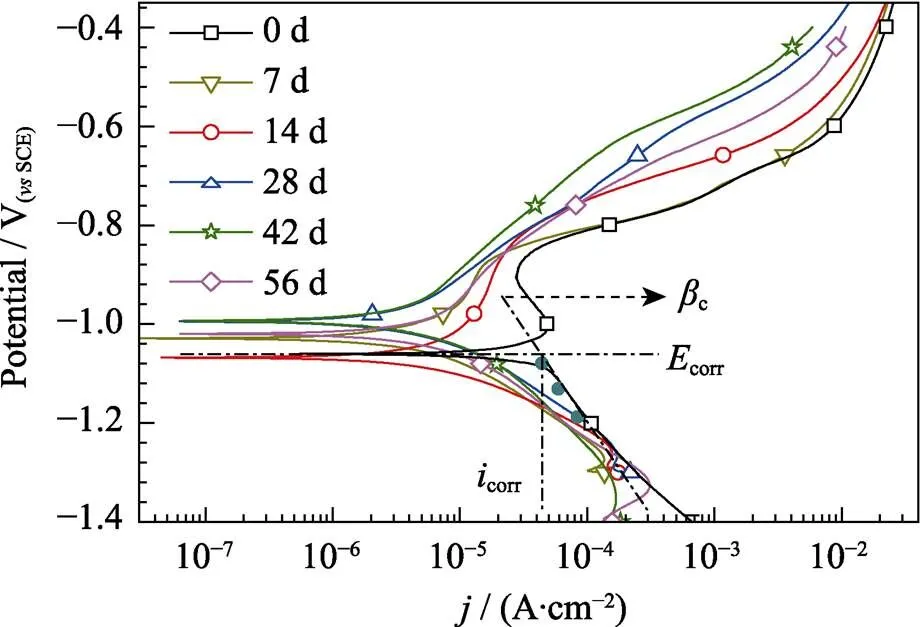
圖1 鋅浸泡在(37±0.5 )℃林格氏液中7、14、28、42和56 d的PDP曲線
根據電化學實驗結果可以看出, 浸泡過程中產生的腐蝕產物層對基體具有一定的保護作用, 顯著降低了鋅的腐蝕速率, 并在整個浸泡過程中保持穩定。通過極化曲線計算所得的鋅在林格氏液中的降解速率要高于臨床上理想的降解速率標準0.02 mm/a[1], 而通過電化學方法估算樣品的腐蝕速率是方法之一, 后面將通過失重法進一步分析腐蝕速率。
鋅在林格氏液中浸泡不同時間后的電化學阻抗譜如圖3所示。從圖3(a)中可以看出, 鋅在浸泡后的阻抗環相較于浸泡前明顯增大。隨著浸泡時間的延長, 樣品阻抗環進一步增大。從阻抗Bode圖和Nyquist圖可以看出, 鋅在浸泡前具有兩個時間常數并在低頻區域出現擴散控制現象, 可能是由于鋅電極表面反應過快, 電極表面附近反應物無法擴散, 因此反應由反應物從溶液本體擴散到電極表面的過程控制[19-20]。而當表面被腐蝕產物覆蓋后, 反應速率下降, 擴散控制現象消失, 取而代之的是中頻區域時間常數的增大, 可能是由于腐蝕產物的分層造成的。因此, 鋅在浸泡前的阻抗曲線可以用電路圖s(p1(p1(dl(ctw))))[15](圖4(a))進行擬合分析。鋅在浸泡過程中的阻抗曲線可以用電路圖s(p1(p1(p2(p2(dlct))))) (圖4(b))進行擬合分析。其中,s代表溶液電阻;p1、p1和p2、p2分別代表外層和內層腐蝕產物的電容和電阻;dl和ct分別代表雙電層的電容和電阻, 與界面電荷轉移反應有關,ct反映了抵抗腐蝕的程度;w表示擴散電阻。
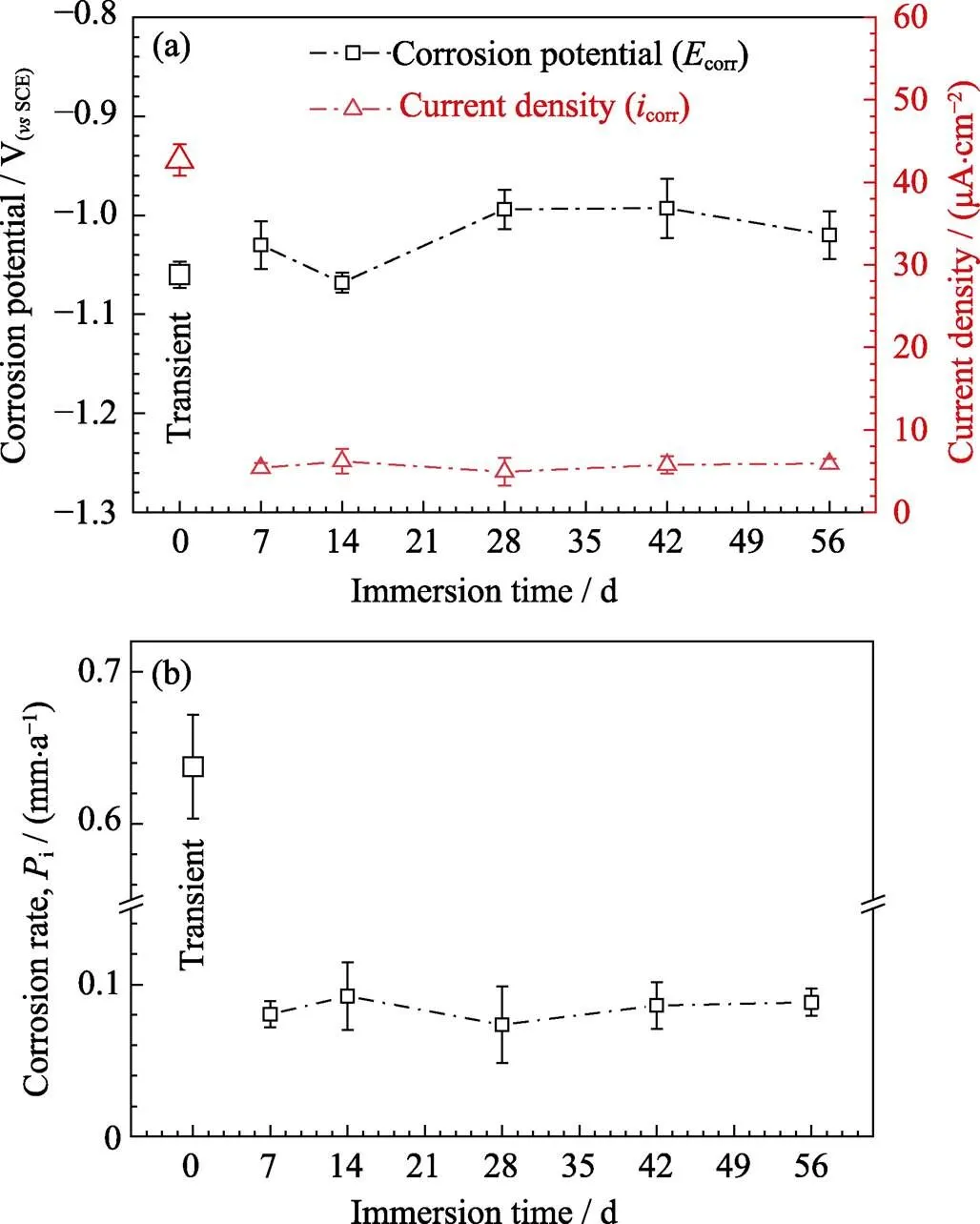
圖2 根據PDP曲線計算的自腐蝕電位Ecorr和自腐蝕電流密度icorr (a)和根據自腐蝕電流密度計算的腐蝕速率Pi (b)
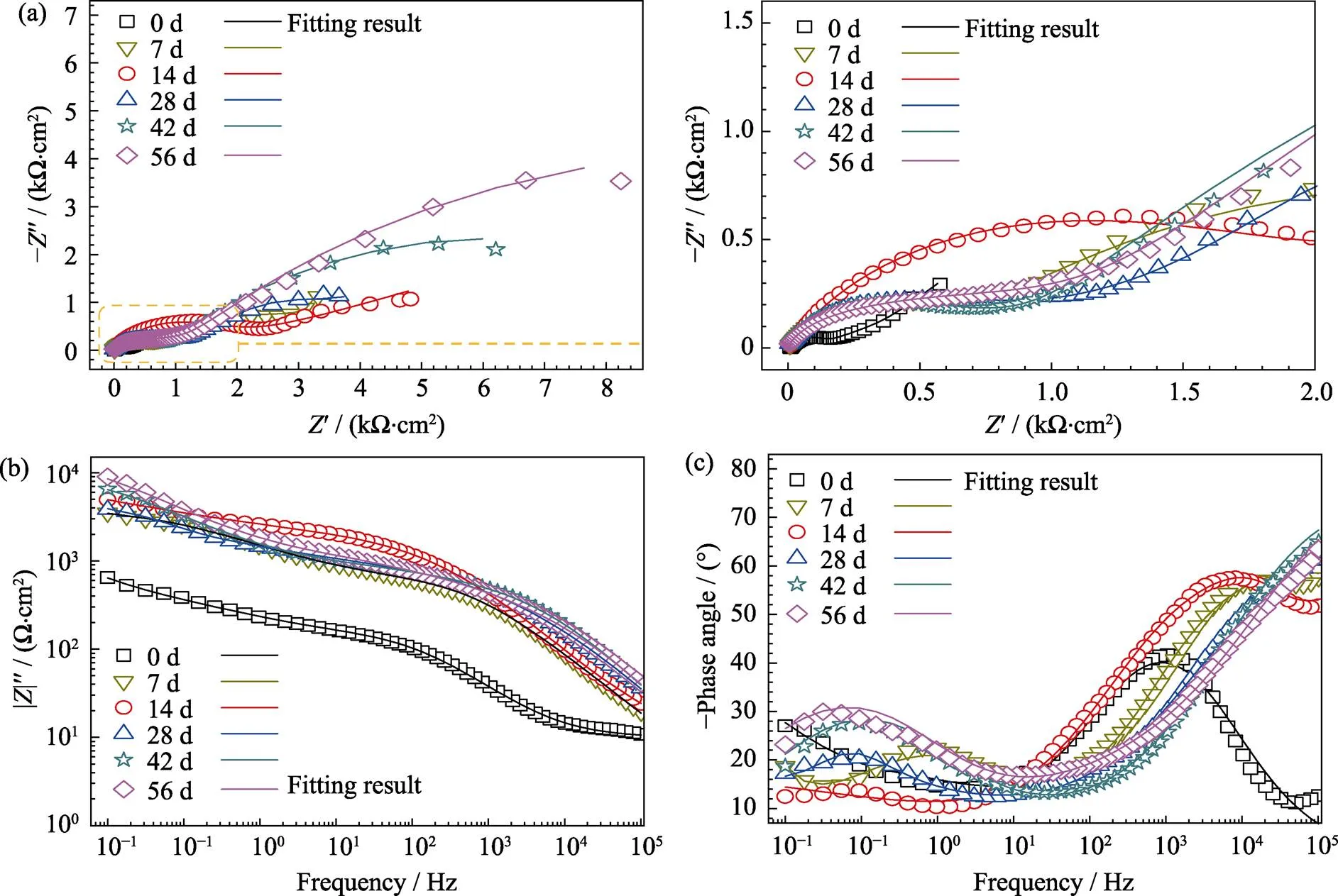
圖3 鋅浸泡在(37±0.5) ℃下的林格氏液中7、14、28、42和56 d后EIS測試結果
根據電路圖擬合出的動力學參數值如表1所示。其中, 腐蝕產物電阻p(p1+p2)和雙電層電阻ct隨浸泡時間的趨勢如圖4(c)所示, 浸泡后腐蝕產物電阻p和雙電層電阻ct均遠高于浸泡前的電阻。隨著浸泡時間的延長, 涂層總電阻未出現明顯的變化, 這一結果與極化計算所得結果相吻合, 說明腐蝕產物層具有良好的穩定性。同時, 根據內外層腐蝕產物層的擬合電阻可以看出, 鋅表面生成的腐蝕產物層是由致密的內層與相對疏松的外層構成。極化電阻polar與雙電層電阻ct可以反映樣品整體的抗腐蝕能力[21], 浸泡前極化電阻可由公式polar=ct+w+p算出, 浸泡后極化電阻可由公式polar=ct+p算出, 隨浸泡時間變化規律如圖4(d)所示。polar與ct規律相似, 隨著浸泡時間的延長, 樣品的腐蝕抵抗能力整體呈增加趨勢, 這主要由于表面腐蝕產物層的保護作用。
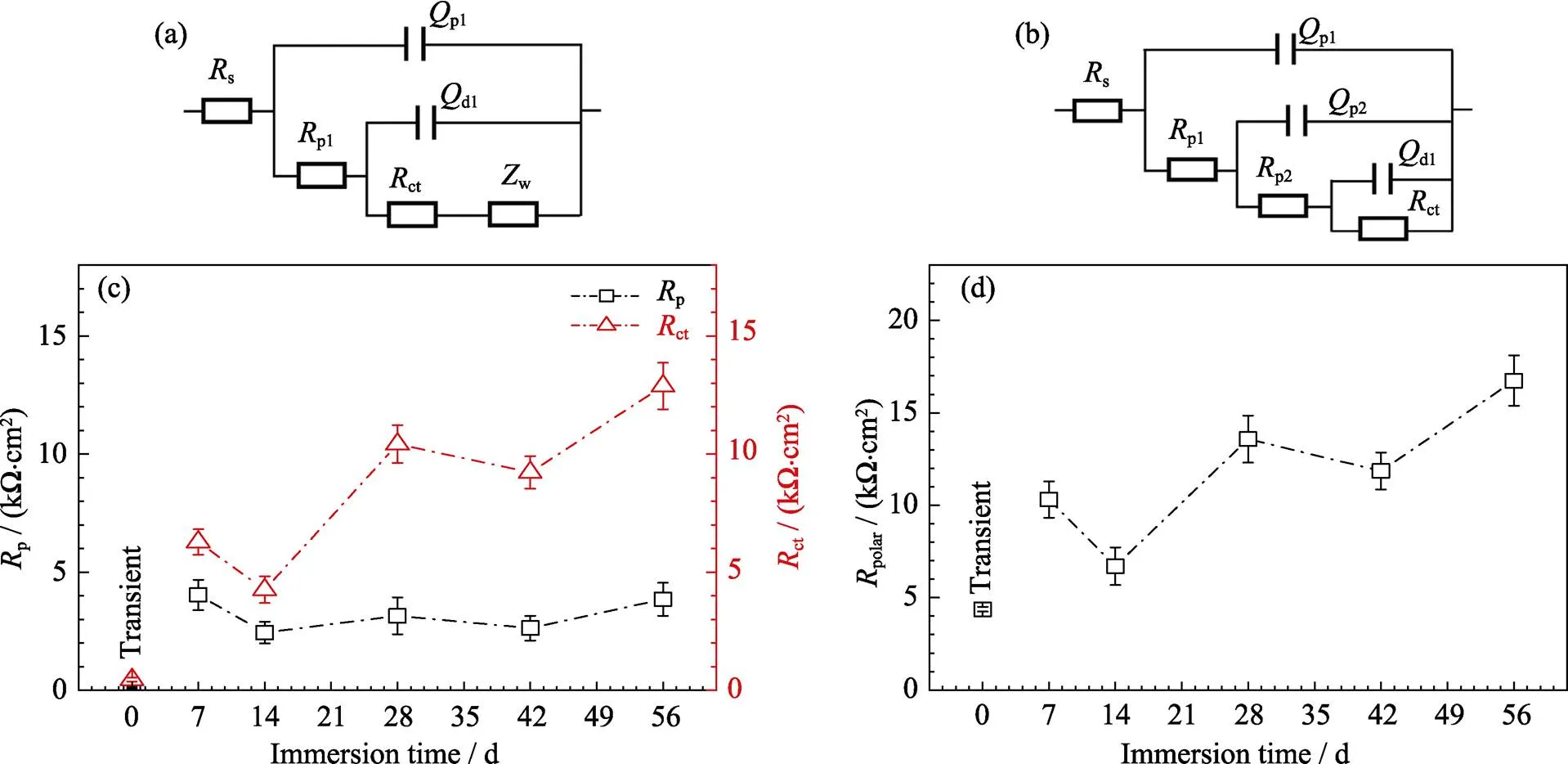
圖4 鋅在浸泡前的EIS數據擬合電路圖(a), 浸泡7~56 d后EIS數據擬合電路圖(b), 界面電荷轉移電阻Rct和腐蝕產物電阻Rp的擬合結果(c), 極化電阻Rpolar的擬合結果(d)
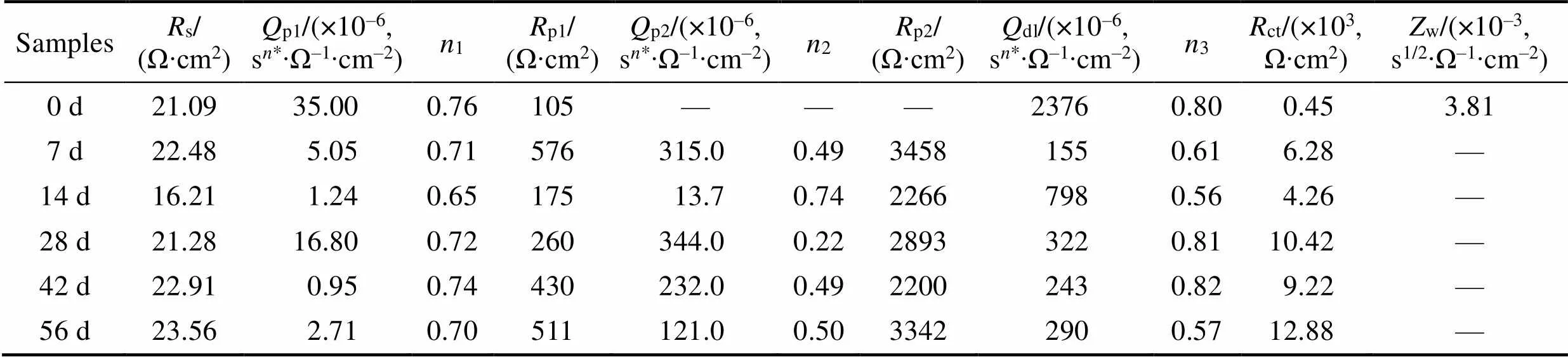
表1 鋅在(37±0.5) ℃林格氏液中的EIS擬合結果
*: s indicates the time unit "second", andindicates diffusion index
2.2 腐蝕產物形貌及截面分析
圖5為鋅在林格氏液中浸泡不同時間后的表面形貌。從體式顯微形貌照片可以看出, 隨著浸泡時間的延長, 表面腐蝕區域逐漸擴展到樣品整個表面, 并伴隨著白色腐蝕產物的積累與粗糙度的增加。整個浸泡過程中未觀察到鋅表面出現嚴重的局部腐蝕以及大量腐蝕產物堆積。從表面SEM照片可以看出, 鋅在浸泡7 d后的表面局部腐蝕產物主要呈細條花棒狀和塊狀。隨著浸泡時間的延長, 表面腐蝕產物逐步變成圓球小顆粒狀和層片狀, 最后逐漸覆蓋住樣品表面。
為了進一步觀察腐蝕產物及被腐蝕產物覆蓋的基體的降解行為, 對浸泡56 d后的樣品進行了截面分析, 如圖6所示。從圖6(a)的截面掃描電鏡照片可知, 腐蝕產物層厚度約為60 μm, 并由表面到基體逐漸變致密。圖6(b)為截面的EDS線掃數據圖, 根據測試結果, 可以明顯地看出腐蝕產物層與基體的分界點, 腐蝕產物層中主要包括Ca、O、C、Zn四種元素, 可以看出元素隨厚度變化的趨勢, 但更精確的表面成分需要結合后續的XPS結果來分析。從截面觀察基體與腐蝕產物的界面層可知, 基體的腐蝕為均勻腐蝕, 未觀察到嚴重局部腐蝕坑。

圖5 鋅浸泡在(37 ± 0.5) ℃的林格氏液中7(a)、14(b)、28(c)、42(d)和56(e) d時的表面形貌
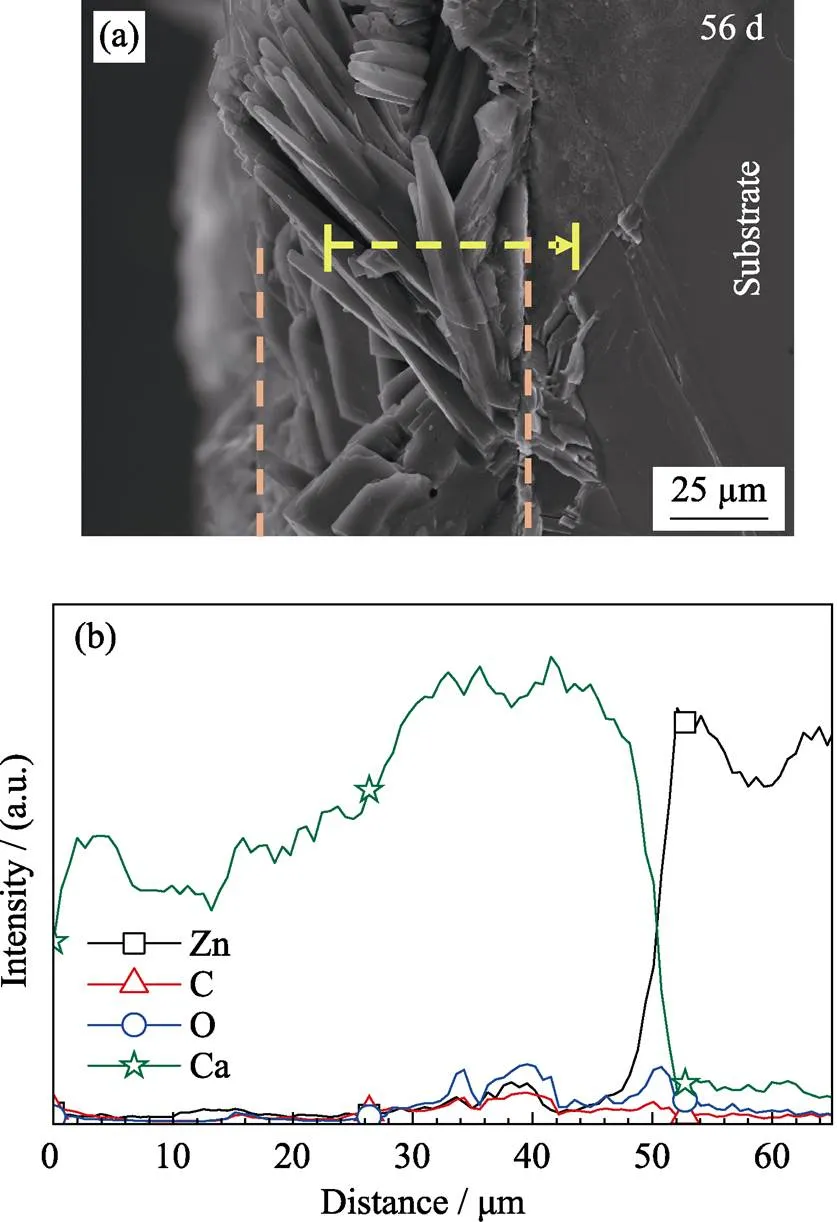
圖6 鋅浸泡在(37±0.5) ℃林格氏液中56 d的截面掃描電鏡照片(a)和截面EDS線掃圖(b)
2.3 腐蝕產物結構及成分分析
圖7為浸泡不同時間后樣品表面腐蝕產物的XRD圖譜。從圖中可以看出腐蝕產物主要為CaCO3和Zn5(CO3)2(OH)6, 并且兩種腐蝕產物的量隨著浸泡時間的延長而增多。這一結果與表面形貌觀察相吻合, 浸泡過程中材料表面首先形成致密的細條狀CaCO3腐蝕產物內層, 對基體起到良好的保護作用。隨著浸泡時間的延長, 圓球狀Zn5(CO3)2(OH)6腐蝕產物逐漸積累, 并與CaCO3混合形成疏松的外層腐蝕產物。
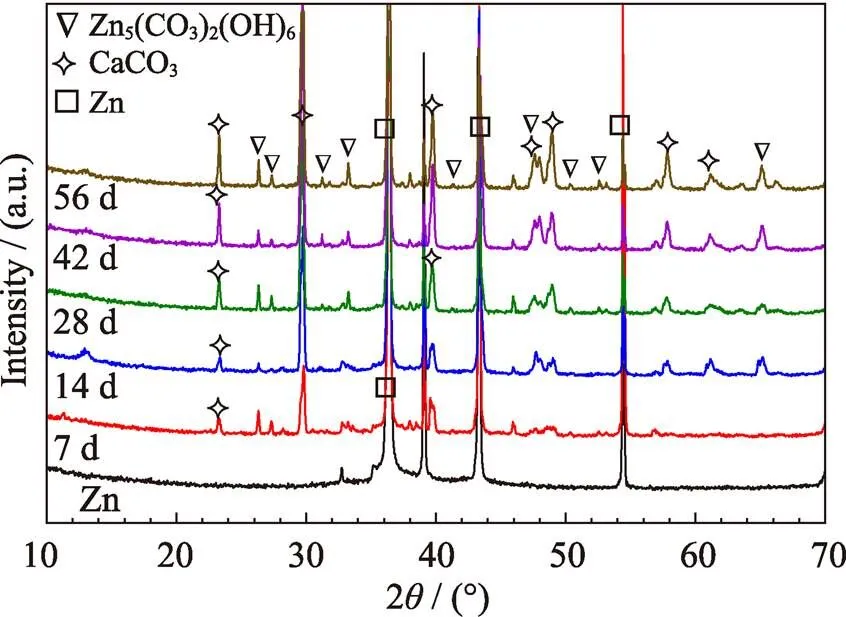
圖7 鋅浸泡在(37±0.5) ℃林格氏液中7、14、28、42和56 d的XRD圖譜
為進一步表征腐蝕產物化學鍵的結合狀態, 對浸泡56 d樣品和純鋅進行了X射線光電子能譜檢測, 如圖8所示。從圖8(a)全譜中觀察到浸泡56 d樣品與純鋅比, 新增了Ca2p的特征峰, Zn2p峰強度明顯降低, C1s峰強度明顯增強, 表明腐蝕產物中Ca、C含量增加。圖8(b)進一步對O1s高分辨進行了分峰處理, 發現浸泡56 d后在 529.1 eV處出現CaCO3的新峰。圖8(c)對Ca2p高分辨進行了分峰處理, 56 d樣品出現了三個特征峰, 分別是350.6 eV出現了CaCO3、Ca2p1/2的峰; 347.2 eV出現了CaCO3、Ca2p3/2的峰; 344.6 eV出現了CaO的峰。故XPS檢測結果進一步證明, 腐蝕產物中含有碳酸鈣鹽等化合物。
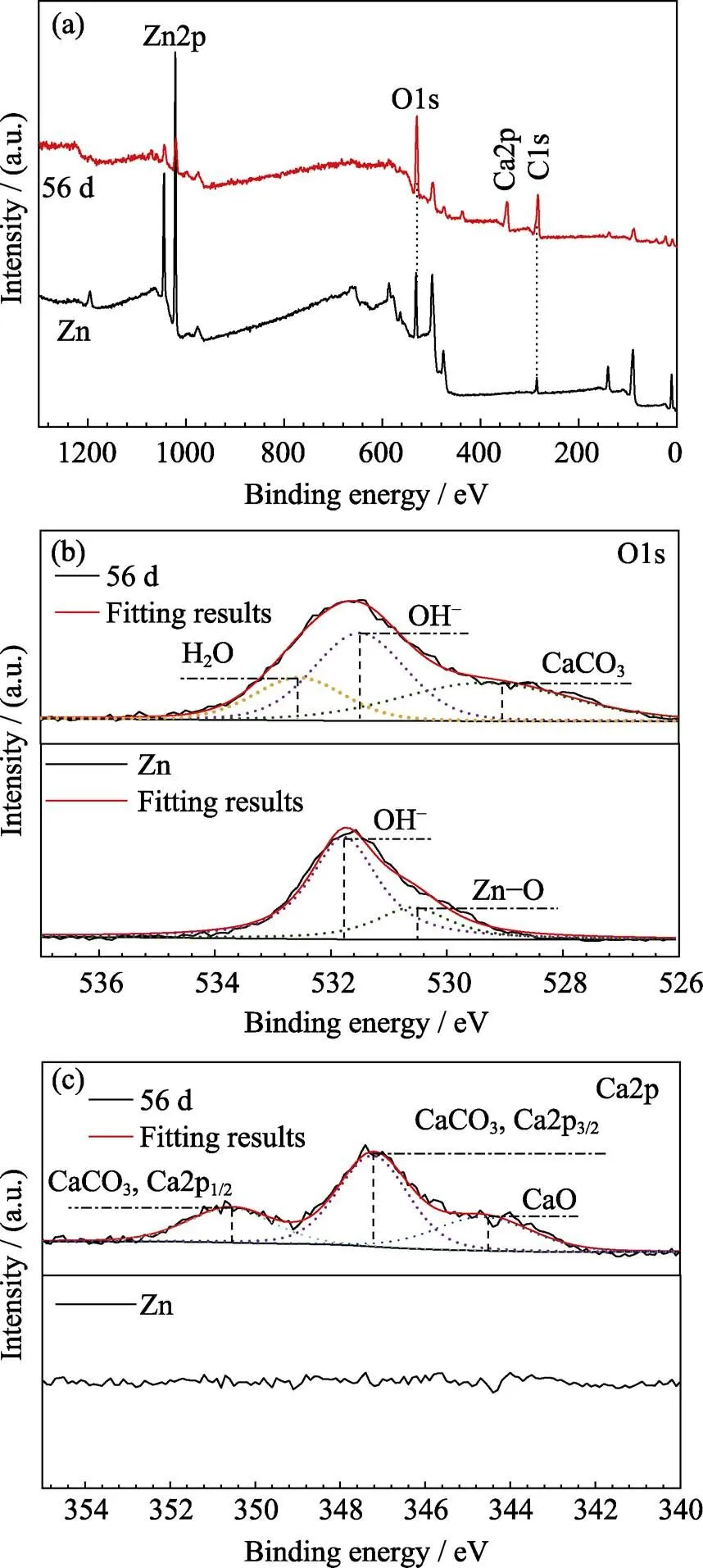
圖8 鋅浸泡在林格氏液56 d后和浸泡前的XPS圖譜
2.4 去除腐蝕產物分析
圖9為鋅在林格氏液中浸泡不同時間后并去除樣品表面腐蝕產物后的SEM照片。從圖中可以看出, 樣品在浸泡7 (圖9(a,a1))和14 d (圖9(b,b1))后, 表面腐蝕較均勻, 腐蝕區域為河流狀形貌, 局部有微小的腐蝕坑。隨著浸泡時間的延長, 表面腐蝕坑逐漸增大, 并在42 d (圖9(d,d1))時出現大約10 μm寬的裂紋。到56 d (圖9(e, e1))時, 裂紋進一步變長加深。這可能由于浸泡前期先在含有雜質(Fe、Ni等)處發生的腐蝕, 隨著浸泡時間的延長, 腐蝕產物逐漸覆蓋腐蝕坑, 到浸泡后期, 基體材料在少量穿透腐蝕產物層的電解液作用下發生電偶腐蝕促進了腐蝕坑的縱向發展, 形成了小孔腐蝕特征[22-23]。圖10為浸泡56 d樣品去掉腐蝕產物后的截面SEM照片, 可以看出樣品為局部腐蝕, 局部有腐蝕溝槽, 在溝槽里面有微小的腐蝕坑。對比去除腐蝕產物前后的樣品, 去除腐蝕產物前表面表現為大體均勻腐蝕, 去除腐蝕產物后基體表現為局部腐蝕, 說明去除腐蝕產物才能更準確地分析基體的腐蝕模式。

圖9 鋅浸泡在(37±0.5) ℃林格氏液中7(a)、14(b)、28(c)、42(d)和56(e)d后去除腐蝕產物的表面形貌
2.5 失重法腐蝕速率分析
圖11為失重法測量浸泡后的腐蝕速率w。鋅浸泡后的腐蝕速率大約為0.3 mm/a到0.5 mm/a之間, 遠遠高于電化學計算所得腐蝕速率, 也顯著高于鋅在其它溶液中的腐蝕速率[11]。相比于根據極化曲線計算出的腐蝕速率i, 在腐蝕速率大小上有差別, 但隨著浸泡時間的變化規律是一致的。兩種方法都比較準確地計算了鋅在林格氏液中的腐蝕速率, 但電化學方法是測試樣品浸泡后暫態的腐蝕速率, 而失重法是測試的整個浸泡過程的腐蝕速率, 故腐蝕速率計算值不同。

圖10 鋅浸泡在(37±0.5) ℃林格氏液中56 d去除腐蝕產物后的截面掃描電鏡照片
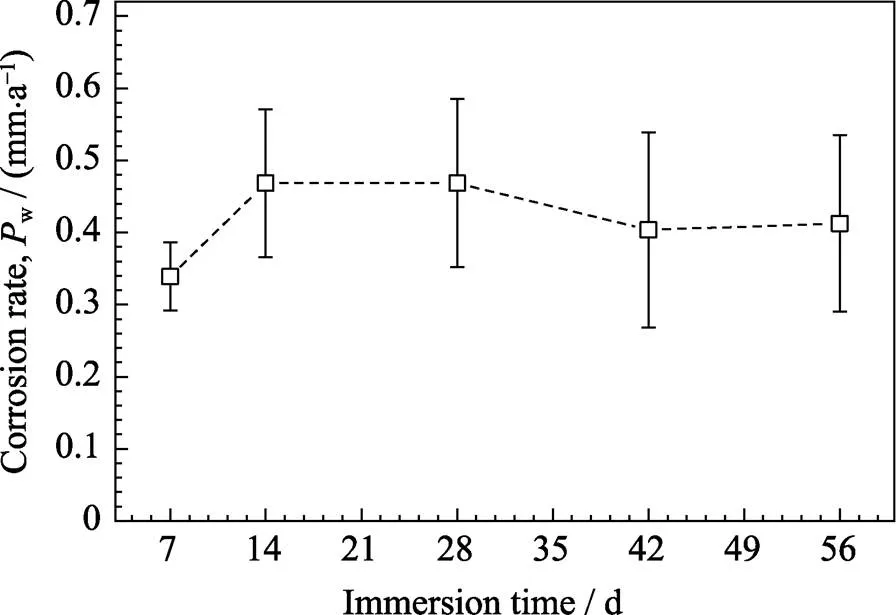
圖11 失重法測量鋅浸泡在(37±0.5) ℃林格氏液中7、14、28、42和56 d的腐蝕速率
3 結論
本研究采用電化學測試、表面形貌及成分分析和去除腐蝕產物等方法, 對生物可降解鋅金屬在林格氏液中的體外腐蝕降解演化行為進行了56 d的長期評價。結果表明, 鋅在林格氏液長期浸泡過程中生成的腐蝕產物層由初期生成的相對致密的條塊狀CaCO3內層及后期生成的較為疏松的CaCO3和Zn5(CO3)2(OH)6混合外層構成, 具有良好的保護作用和穩定性。去除腐蝕產物后發現, 鋅在林格氏液中主要為局部腐蝕, 局部有腐蝕溝槽, 在溝槽里面有微小的腐蝕坑, 且隨著浸泡時間的延長, 腐蝕溝槽增大。鋅在林格氏液浸泡中的腐蝕速率通過失重法計算為0.3~0.5 mm/a, 通過電化學方法測試為0.06到0.10 mm/a, 低于浸泡前的腐蝕速率, 說明浸泡過程中腐蝕產物層對基體有一定的保護作用。
[1] BOWEN PATRICK K, DRELICH JAROSLAW, GOLDMAN JEREMY. Zinc exhibits ideal physiological corrosion behavior for bioabsorbable stents.,2013, 25(18): 2577–2582.
[2] BOWEN PATRICK K, SHEARIER EMILY R, ZHAO SHAN,Biodegradable metals for cardiovascular stents: from clinical concerns to recent Zn-Alloys.,2016, 5(10): 1121–1140.
[3] MOSTAED EHSAN, SIKORA-JASINSKA MALGORZATA, DRELICH JAROSLAW W,Zinc-based alloys for degradable vascular stent applications.,2018, 71: 1–23.
[4] BIN BUM-HO, BHIN JINHYUK, TAKAISHI MIKIRO,Requirement of zinc transporter ZIP10 for epidermal development: implication of the ZIP10–p63 axis in epithelial homeostasis.,2017, 114(46): 12243–12248.
[5] ZHU DONG-HUI, SU YING-CHAO, YOUNG MARCUS L,Biological responses and mechanisms of human bone marrow mesenchymal stem cells to Zn and Mg biomaterials.,2017, 9(33): 27453–27461.
[6] HAASE HAJO, RINK LOTHAR. Multiple impacts of zinc on immune function.,2014, 6(7): 1175–1180.
[7] LIN SONG, WANG QI-LONG, YAN XIN-HAO,Mechanical properties, degradation behaviors and biocompatibility evaluation of a biodegradable Zn-Mg-Cu alloy for cardiovascular implants.,2019, 234: 294–297.
[8] KAFRI ALON, OVADIA SHIRA, GOLDMAN JEREMY,The suitability of Zn-1.3% Fe alloy as a biodegradable implant material.,2018, 8(3): 153.
[9] SHI ZHANG-ZHI, YU JING, LIU XUE-FENG,Effects of Ag, Cu or Ca addition on microstructure and comprehensive properties of biodegradable Zn-0.8 Mn alloy.,2019, 99: 969–978.
[10] ZHENG YU-FENG, WU YUAN-HAO. Revolutionizing metallic biomaterials.,2017, 53(3): 257–297.
[11] CHEN YING-QI, ZHANG WEN-TAI, MAITZ MANFRED F,Comparative corrosion behavior of Zn with Fe and Mg in the course of immersion degradation in phosphate buffered saline.,2016, 111: 541–555.
[12] T?RNE KARIN, LARSSON MARIANN, NORLIN ANNA,Degradation of zinc in saline solutions, plasma, and whole blood.,2016, 104(6): 1141–1151.
[13] ZHAO LI-CHEN, ZHANG ZHE, SONG YU-TING,Mechanical properties andbiodegradation of newly developed porous Zn scaffolds for biomedical applications.,2016, 108: 136–144.
[14] LIU LI-JUN, MENG YAO, DONG CHAO-FANG,Initial formation of corrosion products on pure zinc in simulated body fluid.,2018, 34(12): 2271–2282.
[15] LIU XIAO, YANG HONG-TAO, LIU YANG,Comparative studies on degradation behavior of pure zinc in various simulated body fluids.,2019, 71(4): 1414–1425.
[16] STANDARD ASTM. G102-89, Standard Practice for Calculation of Corrosion Rates and Related Information from Electrochemical Measurements. Annual Book of ASTM Standards, ASTM International, West Conshohocken, PA,2015.
[17] STANDARD ASTM. G31-72. Standard practice for laboratory immersion corrosion testing of metals.Annual Book of ASTM Standards, ASTM International, West Conshohocken, PA,2004.
[18] SHI ZHI-MING, LIU MING, ATRENS ANDREJ. Measurement of the corrosion rate of magnesium alloys using Tafel extrapolation.,2010, 52(2): 579–588.
[19] HUANG JUN. Diffusion impedance of electroactive materials, electrolytic solutions and porous electrodes: Warburg impedance and beyond.,2018, 281: 170–188.
[20] WU J, ZHANG SD, SUN WH,Influence of oxidation related structural defects on localized corrosion in HVAF-sprayed Fe-based metallic coatings.,2018, 335: 205–218.
[21] SHI ZHI-MING, CAO FU-YONG, SONG GUANG-LING,Low apparent valence of Mg during corrosion.,2014, 88: 434–443.
[22] SIM?ES AM, BASTOS AC, FERREIRA MG,Use of SVET and SECM to study the galvanic corrosion of an iron-zinc cell.,2007, 49(2): 726–739.
[23] BLANDA GIUSEPPE, BRUCATO VALERIO, PAVIA FRANCESCO CARFì,Galvanic deposition and characterization of brushite/ hydroxyapatite coatings on 316L stainless steel.,2016, 64: 93–101.
Long-termCorrosion Behavior of Zinc in Ringer’s Solution
TANG Shuai, ZHANG Wentai, QIAN Junyu, XIAN Peng, MO Xiaoshan, HUANG Nan, WAN Guojiang
(Key Laboratory of Advanced Technologies of Materials, Ministry of Education, School of Materials Science and Engineering, Southwest Jiaotong University, Chengdu 610031, China)
In recent years, zinc-based biodegradable materials have gained significant attention due to their desirable biodegradation rate compared with other extensively explored biodegradable metals, such as magnesium and iron. However, the long-term corrosion behavior of zinc in simulated body fluid remain unclear. In this study, we performed a 56 d immersion test to reveal the long-term evolution of corrosion behavior of zinc in Ringer’s solution using electrochemical methods and surface analysis. The results showed that the corrosion rate of Zn calculated from current density ranged from 0.06 to 0.1 mm/a during the immersion.Its corrosion rate, determined by weight loss method, was from 0.3 mm/a to 0.5 mm/a. The corrosion products were mainly composed of Zn5(CO3)2(OH)6and CaCO3. These products were firm, rod- and block-like formed on Zn surface, and gradually accumulated with increase of immersion time. Its surface morphology after removing corrosion products exhibited increasing sizes of corrosion pits and grooves with increase of immersion time. And width of the corrosion pits and grooves was about 10 μm after 42 d immersion. This study provides a guideline for the further surface modification and biomedical applications of Zn-based materials in terms of biodegradation profile.
biodegradable zinc; long-term corrosion behavior; electrochemical test; surface chemistry
TG178; R318
A
1000-324X(2020)04-0461-08
10.15541/jim20190176
2019-04-25;
2019-06-07
國家重點研發計劃(2016YFC1102500); 國家自然科學基金(21473138)
National Key Research and Development Program of China (2016YFC1102500); National Natural Science Foundation of China (21473138)
唐帥(1992–), 男, 碩士研究生. E-mail: tangshuaicc@163.com
TANG Shuai (1992–), male, Master candidate. E-mail: tangshuaicc@163.com
萬國江, 教授. E-mail: guojiang.wan@home.swjtu.edu.cn
WAN Guojiang, professor. E-mail: guojiang.wan@home.swjtu.edu.cn

- 無機材料學報的其它文章
- BaTiO3-ZnNb2O6陶瓷介電及儲能性能研究
- 高強度預應力陶瓷的發展與探索
- Synthesis, Crystal Structure, and Electrical Conductivity of Pd-intercalated NbSe2
- SnS2 Nanoplates: Synthesis and NO2 Sensing Property
- High Efficient Carbon Quantum Dots/BiOCl Nanocomposite for Photocatalytic Pollutant Degradation
- Removal of Volatile Organic Compounds Driven by Platinum Supported on Amorphous Phosphated Titanium Oxide