強直性肌營養不良的臨床特征、肌電圖及肌肉病理研究
2020-05-08 01:35:48曹秉振呂涌濤胡懷強趙修敏馮肖亞克丙申
臨床薈萃 2020年4期
劉 磊,陳 燕,曹秉振,呂涌濤,胡懷強,趙修敏,馮肖亞,克丙申,郇 英
(1.山東省立第三醫院 神經內科,山東 濟南 250031;2.解放軍第960醫院 神經內科,山東 濟南 250031)
強直性肌營養不良(myotonic dystrophy,DM)是一種常見的常染色體顯性遺傳病,臨床上主要表現為肌強直、肌無力和肌萎縮,常伴有白內障、心律失常、糖尿病、禿發、多汗、性功能障礙和智力減退等全身多系統受累的表現。根據發病年齡不同,臨床上將DM分為成年型DM和先天型DM。通常成年型發病年齡為20~25歲,癥狀相對較輕,預后較好,具有明顯的遺傳早現現象;先天型患兒,在出生時即發病,病情往往極重,常常在短時間內死亡,很少能活過35歲。此外,根據肌強直及肌萎縮累及的部位,又將其分為經典型DM和主要累及肢體近端及肢帶肌的近端強直性肌病(PROMM)或稱為近端強直肌性營養不良(PMD)。PROMM中樞神經系統往往很少受累。DM在全球發病率約為1/8000,但DM在世界各地的發病率不一,DM在日本的發病率接近1:10萬,在歐洲為(3~15):10萬[1],臺灣的研究報告表明,臺灣DM發病率為0.46/10萬[2]。
兩型強直性肌營養不良患者之間的臨床癥狀及體征非常相似,均可出現多系統受損的臨床表現,如:肌強直、肌無力、肌萎縮、禿頂、眼部損害(白內障)及內分泌的改變,實驗室檢查可見肌酶輕度增高或正常,肌電圖可見特征性肌強直電位,部分患者可見周圍神經受損,肌肉活檢顯示肌纖維的肥大及萎縮,肌細胞變性,Ⅰ型和Ⅱ型肌纖維均可受累,但在不同兩型DM患者,可表現為某一種類型的肌纖維損害為主。二者臨床表現也有不同點:在發病年齡上,DM1存在先天性發病,并常伴有嚴重智力障礙,DM2至今沒有先天性發病的報道,且DM2極少損害中樞神經系統;DM1肌萎縮常較嚴重,而DM2肌萎縮少見;DM2常見的臨床表現為肌肥大和肌肉疼痛,肌無力表現為近端或遠端無力,DM1的肌肥大及肌肉疼痛少見,肌無力以遠端為主。
1 臨床資料
共收集2008-2011年在解放軍第960醫院神經內科的住院臨床診斷為DM的患者5例,其中2例來自山東省梁山縣同一家系,家系內3代9人中有2例發病。診斷標準:①中青年起病,慢性病程;②臨床表現為肌強直、肌無力、肌萎縮;③肌電圖可見強直電位;④肌肉病理符合強直性肌營養不良。
患者1:溫某,女,32歲,因“進行性四肢無力、活動不靈10年”于2009年12月2日住院。患者于10年前無明顯誘因出現雙手無力、抓握困難,雙手握緊拳后松開困難,遇冷時上述癥狀加重,天熱時癥狀減輕。8年前出現雙下肢無力,上樓抬腿費力,雙手握拳后伸直費力較前明顯加重,并出現抬頭費力,未行治療,病情進行性加重,近2年患者出現走路容易被絆倒,四肢肌肉有肉跳,咀嚼費力,視力有所減退,并出現月經不規律。曾到石家莊等多家醫院就診,給予激素治療后上述癥狀有所好轉,停用激素后上述癥狀再次加重。既往體健。家系內3代共9人(圖1),姐妹兩人發病,發病年齡均為22歲。入院查體:心肺腹體檢無異常。專科查體陽性體征:雙眼閉合力稍弱,雙側胸鎖乳突肌萎縮,雙上肢近端肌力Ⅴ-級,握力Ⅳ級,雙手背屈肌力為Ⅲ級,跖屈正常。雙下肢近端肌力Ⅴ-級,遠端背屈Ⅱ級,跖屈Ⅳ+級,雙側腱反射對稱性減弱(+)。實驗室檢查:肌酶:α-羥丁酸脫氫酶:201 U/L;肌酸激酶522 U/L;肌酸激酶同工酶:12 U/L血糖:5.3 mmol/L,大致正常心電圖。血常規、肝功能、腎功能、離子、血脂、血糖、凝血、肌鈣蛋白、尿常規、大便常規、感篩未見明顯異常。肌電圖:所檢肌肉以短時限、低波幅、多相波電位為主,提示肌源性損害并見肌強直電位,腓神經運動神經傳導誘發電位波幅降低。不排除肌強直性肌營養不良(圖2、3)。肌肉病理:左脛神經肌肉活檢所示HE染色:光鏡下示肌纖維大小明顯不等,以肥大肌纖維為主,少量散在萎縮肌組織,壞死肌纖維少見,再生肌纖維可見,內核肌纖維明顯增多,占80%~90%,縱切面可見核鏈形成。未見肌漿塊形成,肌間隙增寬,結締組織贈生明顯,未見淋巴細胞浸潤,直管結構及形態正常;MGT染色未見破碎紅纖維和鑲邊空泡。NADH-RT染色兩型肌纖維分辨不清(圖4)。5例患者臨床特征,見表1。
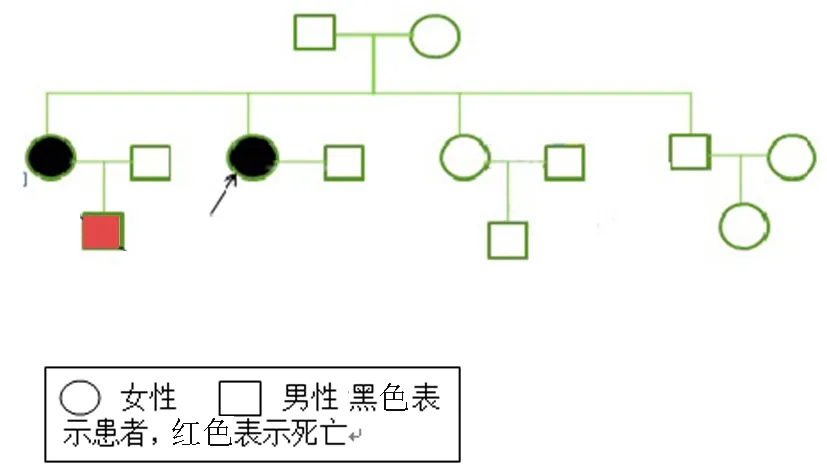
圖1 家系1的遺傳家系圖。紅色患者為該家系的先證者
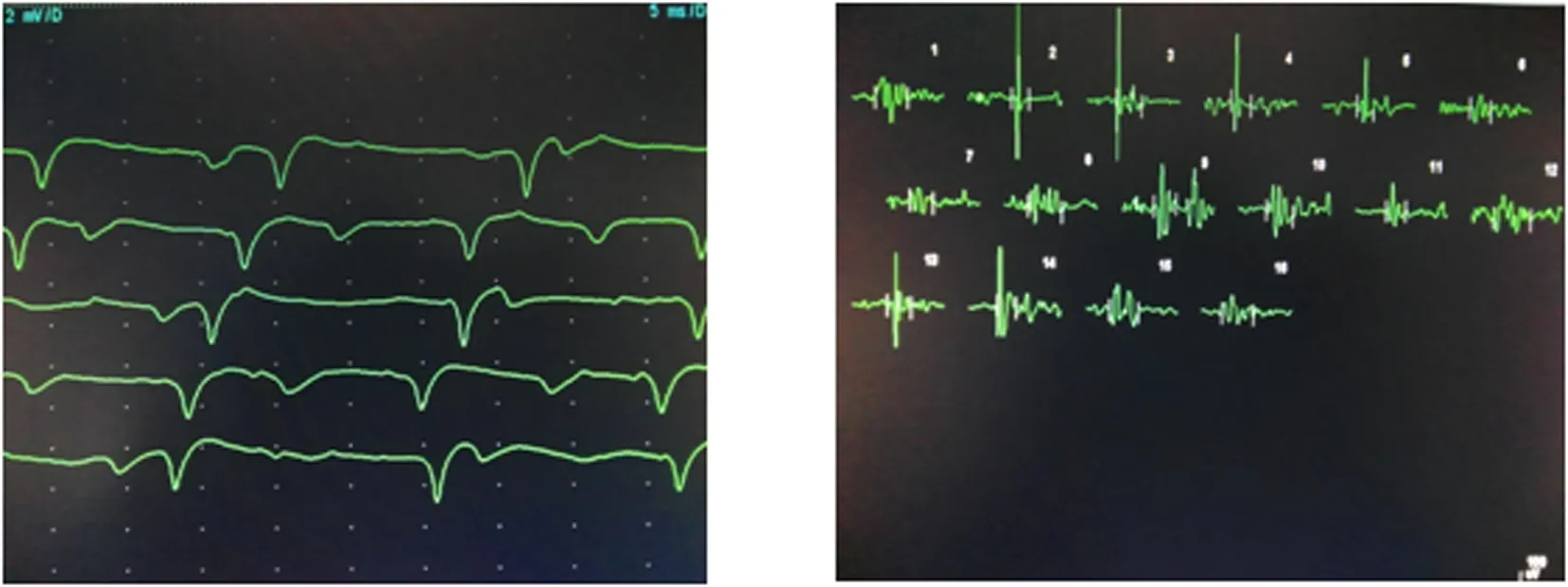
圖2 患者1的肌電圖可見肌強直電位 圖3 患者1肱二頭肌短時限、低波幅、多相波電位為主
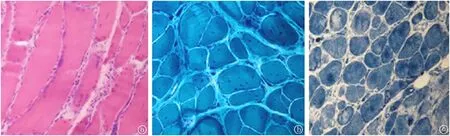
圖4 患者1的左脛神經肌肉活檢a.HE染色(×400):光鏡下示肌纖維大小明顯不等,以肥大肌纖維為主,少量散在萎縮肌組織,壞死肌纖維少見,再生肌纖維可見,內核肌纖維明顯增多,占80%~90%,縱切面可見核鏈形成。未見肌漿塊形成,肌間隙增寬,結締組織贈生明顯,未見淋巴細胞浸潤,直管結構及形態正常。b.MGT染色(×400):未見破碎紅纖維和鑲邊空泡。c.NADH-RT染色(×400):兩型肌纖維分辨不清。
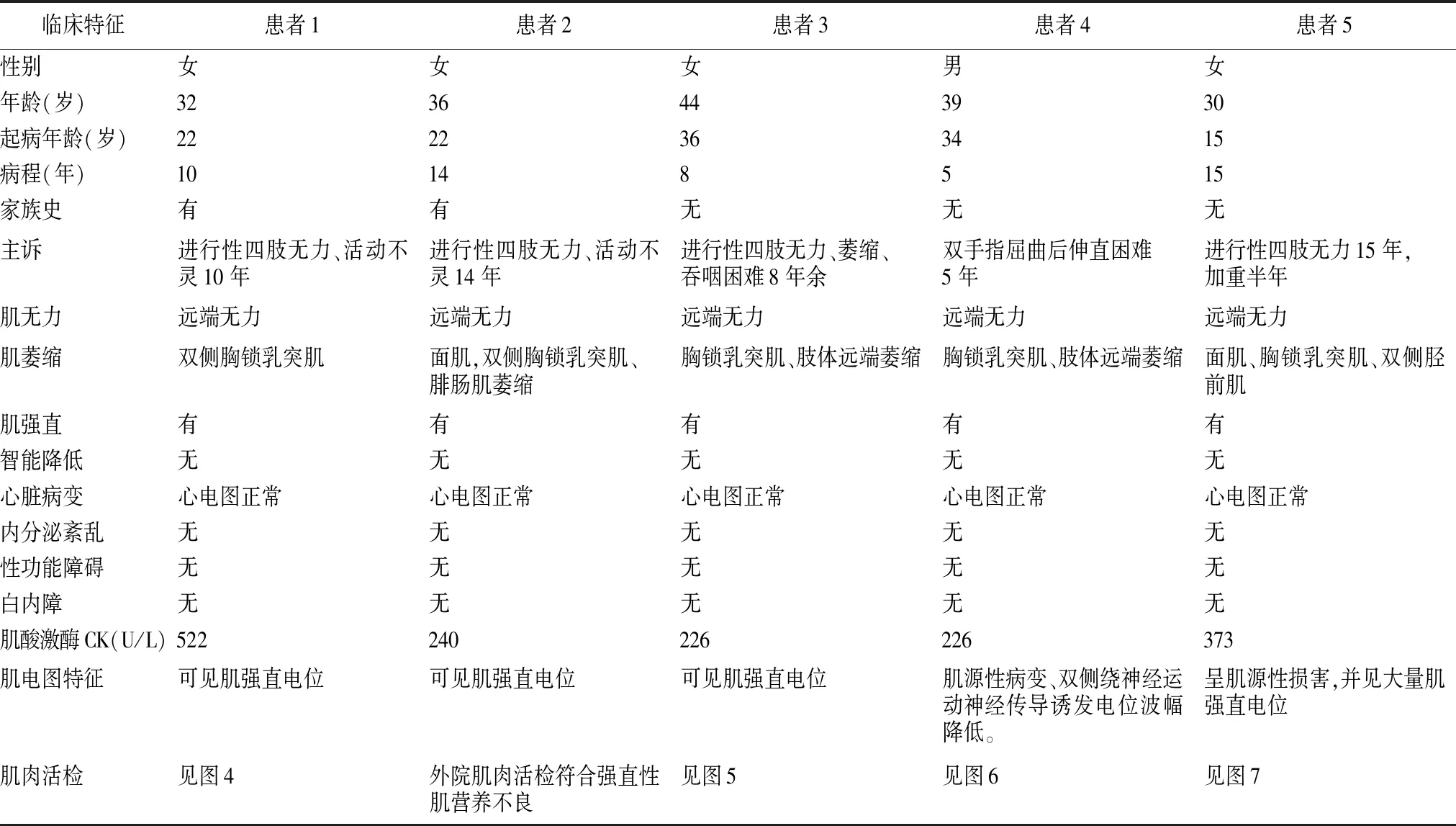
表1 5例患者臨床特征
圖5 患者3的肌肉活檢a.HE染色(×400):肌纖維明顯大小不等,散在萎縮及少數輕度肥大,無變性壞死肌纖維,肌膜核增多,內膜核多見,未見肌漿塊,未見炎性細胞浸潤,血管結構正常。b.MGT染色(×400):未見破碎紅纖維和鑲邊空泡纖維。c.NADH染色(×400):兩型肌纖維比例大致正常,但1型纖維變小萎縮,肌原纖維網紊亂,可見酶缺失或蟲蝕現象,2型纖維大致正常,少量肥大。
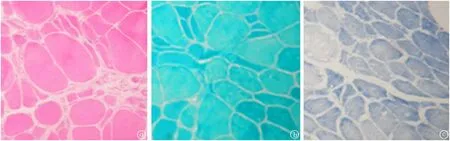
圖6 患者4的左肱二頭肌活檢a.HE染色(×400):肌纖維明顯大小不等,呈雙峰分布。散在萎縮和小群萎縮,肌纖維形態不一,有變鈍變圓的,有角形和不規則的,肥大肌纖維比例多于萎縮肌纖維。每束肌纖維病變程度不同,可見不透明肌纖維,個別肌纖維有空泡樣改變,未見肌纖維壞死,再生和劈裂肌纖維,未見核內移、肌膜核增多現象和肌漿塊。個別肌束肌內膜下結締組織輕度贈生,肌間質增寬,間質小血管未見明顯異常。b.MGT染色(×400):未見破碎紅纖維和鑲邊空泡。c.NADH染色(×400):以選擇性1型肌纖維萎縮為主,2型肌纖維肥大改變,無明顯肌原纖維網紊亂。
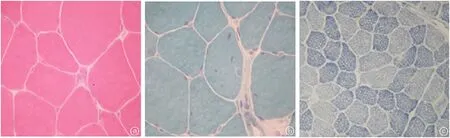
圖7 患者5的肌肉組織活檢a.HE染色(×400):光鏡下顯示肌纖維大小輕度不等,散在少量萎縮纖維,可見不透明纖維,肌纖維肥大不明顯,核內移多見,未見肌漿塊,無明顯結締組織增生。b.MGT染色(×400):未見RRF和鑲邊空泡纖維,血管結構及形態正常,無炎性細胞浸潤。c.NADH-RT染色(×400):顯示兩型纖維比例正常,少量部分1型纖維變鈍變小。
2 討 論
強直性肌營養不良是一種以肌無力、肌萎縮、肌強直為主要特點的常染色體顯性遺傳疾病,常伴隨白內障、心律失常、糖尿病、禿發、多汗、性功能障礙和智力減退等多系統受累的臨床表現。目前在分子生物學水平上將其可分為DM1和DM2[3]。兩者臨床表現具有共同點,都表現為肌強直、肌無力、肌萎縮,也具不同點,如DM1成年型患者平均發病年齡20~40歲;DM2發病年齡在13~67歲,中位年齡48歲。
兩型DM的臨床表現較相似[4]:①肌強直、肌無力、肌萎縮;②面肌、屈勁肌、肢體遠近端肌肉均可受累;③眼部受累,主要表現為白內障;④心臟受累,主要表現心律不齊及傳導阻滯;⑤男性性功能低下、睪丸萎縮;⑥糖尿病或糖耐量異常;⑦低γ丙球蛋白血癥,肌酸激酶和卵泡刺激素水平增高[5]。兩者不相同的是,DM2主要臨床表現為肌痛[6],肌萎縮少見且程度較輕,不損傷中樞神經系統,常見腓腸肌肥大。至今未發現DM2先天性患者,及類似DM1患者那樣嚴重的中樞神經系統損害病例報告[7]。
目前診斷DM的標準[8]主要為:①青中年起病,進展緩慢;②常有明顯的家族史;③臨床表現為肌強直、肌萎縮、肌無力,肌萎縮主要以面部、頸部、肢體遠端肌肉為主,伸肌重于屈肌;④全身多系統受損,其中白內障、男性禿頂是本病的突出體征;⑤肌電圖表現為特征性的肌強直電位和肌源性損害;⑥肌肉活檢顯示主要的肌肉病理為核內移、核鏈形成、肌萎縮以Ⅰ型纖維為主。一般根據以上臨床和病理方面的特點進行診斷,但是對輕型或不典型病例則需借助分子生物學方法進行基因診斷,直接測定DMPK基因CTG重復序列的長短已經成為確診DM1的依據。
本次共收集5例患者,男1例,女4例,發病年齡為15~36歲,中位年齡為22歲,其中2例來自同一家系,家系內其他成員均無類似臨床表現,且2例均未有子女,家族遺傳現象不明確。5例均以肌無力為首發癥狀,并伴有肌萎縮、肌強直,病程為5~15年,平均10.4年,均存在肌萎縮,主要表現為面肌、胸鎖乳突肌及肢體遠端肌肉萎縮,5例患者均接受心電圖檢查,且心電圖均正常,眼科檢查均未見患有白內障,同一家系內2例患者均為女性,1例未生育,1例生有1子,但出生后即夭折,表明該家系內患者生殖系統受累,肌電圖檢查5例均為肌源性損害,均可見肌強直電位,有3例可見周圍神經波幅降低,提示周圍神經損害。
已經證明,DM的典型骨骼肌病理改變為肌纖維大小不一,形態成角形、圓形和不規則形;有大量核內移纖維,伴較多的肌纖維肥大、增殖和分裂,但無明顯的肌纖維壞死,萎縮肌纖維可出現明顯的肌漿塊為本病的特點,但并非每個患者都出現,ATP酶染色顯示肌纖維萎縮以Ⅰ型為主,Ⅱ型肌纖維肥大,可有肌源性群組化現象[9]。
本次觀察的5例患者4例在我科行肌肉活檢術,1例為院外檢查,可見肌纖維明顯大小不等。存在萎縮和肥大,肌萎縮主要表現為Ⅰ型肌纖維,肌纖維形態不一,有變鈍變圓的,有角形和不規則的,肥大肌纖維比例多于萎縮肌纖維。每束肌纖維病變程度不同,可見不透明肌纖維,個別肌纖維有空泡樣改變,壞死肌纖維少見,再生肌纖維可見,內核肌纖維明顯增多。1例患者病理縱切面可見核鏈形成。5例患者均未見肌漿塊形成,研究顯示肌漿塊陽性率不足50%,所以肌肉活檢未見肌漿塊不能排除DM的診斷。綜上所述,5例患者均表現為肌纖維萎縮及肥大,且部分患者可見肌漿塊及核鏈,病理均符合強直性肌營養不良的診斷。強直性肌營養不良的骨骼肌病理改變,對本病的診斷和鑒別診斷有重要的臨床意義。
基因檢測可以對 DM 患者明確診斷和分型,根據其致病基因位點的不同分為兩種類型:DM1型為染色體19q13.3的DMPK基因3’端非翻譯區三核苷酸CTG序列異常重復擴增所致和DM2型為染色體3q21.3的ZNF9基因第一內含子中四核苷酸CCTG重復序列的異常擴增所致。DM的發病機制尚不清楚,目前公認的學說為一種獲得性RNA毒性機制致病,異常擴增的CUG或CCUG在細胞核中異常聚集,影響剪切因子水平,進而導致多種mRNA剪切異常,引起多系統損害的臨床表現[10-11],文獻報道,發現CTG的重復數目與肌肉病理核內移纖維比例呈正相關,即CTG的重復個數越多,肌肉病理核內移受累的程度越重;這與Brook等[12]的研究結果相一致。側面反映肌肉病理診斷的重要性。強直性肌營養不良癥通過基因檢測可以明確該病的分型診斷,但可能存在未知的致病基因[13-14]。本組患者未行基因檢測,待隨訪后完善基因檢測。因此在臨床工作中需仔細追問病史,結合患者的肌電圖及肌肉病理結果予以診斷,注意與先天性肌強直、先天性副肌強直、面肩-肱型肌營養不良癥鑒別。確診仍需基因診斷。
治療上:大多數患者可以通過防止各種刺激尤其是寒冷和過度運動,能夠保持很好的生活質量。美西律可用于改善肌強直癥狀。對于以肌無力為主要癥狀的患者可以添加噻嗪類利尿劑。隨著對強直性肌營養不良分子機制的研究,現已提出更為有效 、特異的新治療方法。靶向分子治療尤其是反義寡核苷酸(ASO)治療于體外、動物模型中都已經取得了成功有研究給予強直性肌營養不良Ⅰ型轉基因小鼠全身給予ASO,快速的敲除了骨骼肌中 CUG(exp)RNA,糾正該病的生理學、組織病理學、轉錄組學特點,且用藥結束后藥效持續了1個小時[15]。但在人體試驗中如何在無毒的劑量下 ,準確的選定受累組織仍然是一個難題。強直性肌營養不良Ⅰ型的早期死亡70%是由心肺并發癥引起,積極的監控及及時治療可有效的減低病死率及患病率。
