Metabolic and genetic assessments interpret unexplained aggressive pulmonary hypertension induced by methylmalonic acidemia: A case report
2020-04-25 05:00:46HongYuLiaoXiaoQingShiYiFeiLi
World Journal of Clinical Cases 2020年5期
Hong-Yu Liao,Xiao-Qing Shi,Yi-Fei Li
Hong-Yu Liao,Xiao-Qing Shi,Yi-Fei Li,Department of Pediatrics and Ministry of Education Key Laboratory of Women and Children's Diseases and Birth Defects,West China Second University Hospital,Sichuan University,Chengdu 610041,Sichuan Province,China
Abstract
BACKGROUND
Pulmonary hypertension(PH) causes significant morbidity and mortality in diverse childhood diseases.However,limited information has been reported to obtain a good understanding of pediatric PH.Gaps exist between genome sequencing and metabolic assessments and lead to misinterpretations of the complicated symptoms of PH.Here,we report a rare case of a patient who presented with severe PH as the first manifestation without significant cardiovascular malformation and was finally diagnosed with methylmalonic aciduria(MMA) after metabolic and genomic assessments.
CASE SUMMARY
An 11-year-old female presented with an aggressive reduction in activity capability and shortness of breath for only 4 mo and suffered from unexplained PH.A series of examinations was performed to evaluate any possible malformations or abnormalities of the cardiovascular system and lungs,but negative results were obtained.The blood tests were normal except for manifestations of microcytic anemia and elevated total homocysteine.Computed tomography and magnetic resonance imaging failed to identify any pulmonary diseases.Cardiac catheterization examination identified a small right coronary artery to pulmonary artery shunt and severe PH.During the follow-up,PH progressed rapidly.Then,genome sequencing and metabolic disorder screening were performed,which confirmed a diagnosis of MMA with MMACHC c.80A >G/c and 609G > A mutations.Vitamin B12,betaine and bosentan were then administered as the main treatments.During the 6-mo follow-up,the pulmonary artery pressure dropped to 45 mmHg,while the right ventricle structure recovered.The patient's heart function recovered to NYHA class II.Metabolic disorder analysis failed to identify significant abnormalities.
CONCLUSION
As emerging types of metabolic dysfunction have been shown to present as the first manifestation of PH,and taking advantage of next generation sequencing technology,genome sequencing and metabolic disorder screening are recommended to have a more superior role when attempting to understand unclear or aggressive PH.
Key words:Pulmonary hypertension;Methylmalonic acidemia;Genomic sequence;Metabolic disorder;Case report
INTRODUCTION
Pulmonary hypertension(PH) is a progressive disease that causes significant morbidity and mortality in various childhood diseases[1].In childhood,abnormal heart structures,especially left-to-right shunt congenital heart diseases,are the main causes of PH[2].Despite the availability of new medications,the long-term outcomes are still poor[3].Recently,emerging studies have reported that genetic and metabolic assessments have been performed to demonstrate the molecular basis of unexplained PH.However,it is unknown which patients need to receive a genomic and metabolic assessment.Here,we report a rare case of severe PH as the first manifestation in a patient who underwent a delayed genomic and metabolic assessment to accurately understand the disorder.In addition,we reconsidered the criteria for unexplained PH that indicate the need for a combined genomic and metabolic assessments.
CASE PRESENTATION
Chief complaints
An 11-year-old girl complained of an aggressive reduction in activity capability and shortness of breath and presented to our cardiovascular department.
History of illness
The patient started to demonstrate a reduction in activity capability 4 mo ago and progressed with accelerated worsening of her condition within the most recent 1 mo,presenting severe shortness of breath.However,the patient denied any history of past illness.
Physical,laboratory and imaging examinations
An enhanced P2 sound and a systolic murmur between sternal ribs 2 and 3 wereobserved.She suffered from NYHA class II heart function.Echocardiography revealed a large right ventricle and pulmonary artery trunk,with an estimated 60 mmHg pulmonary arterial pressure and normal left heart function.Computed tomography and magnetic resonance imaging scans excluded lung disease and cardiomyopathy,which could lead to PH.In addition,the results of the autoimmune antibody analysis were negative and excluded any connective tissue or rheumatological diseases.
Cardiac catheterization examination
The right ventricular catheter examination showed an elevated right ventricular pressure of 50/2(23) mmHg,main pulmonary artery pressure of 57/21(55) mmHg,left pulmonary artery pressure of 50/29(36) mmHg and right pulmonary artery pressure of 60/31(45) mmHg with a total pulmonary resistance of 8.72 woods,Qp 6.3 L/min.Therefore,the catheter evaluation failed to identify a clear cause.
Further diagnostic work-up
Because of the rapid progression of pulmonary artery pressure with microcytic anemia and elevated homocysteine,metabolic disorders were suspected.Therefore,metabolic screening and genome sequencing was performed for this patient.Metabolic screening showed an elevation of methylmalonic acid of 51.82 μg/L,and two mutations c.80A > G/c and 609G > A were recognized at the gene for methylmalonic aciduria and homocystinuria type C protein(MMACHC),and these mutations were reported to be associated with methylmalonic acidemia,cobalamin C type.
FINAL DIAGNOSIS
This patient was finally diagnosed with methylmalonic acidemia with aggressive PH as the first clinical manifestation.
TREATMENT AND FOLLOW-UP OUTCOME
As metabolic and genome screening need almost 1 mo to be completed,we provided bosentan(endothelin-1 inhibitor) to this patient and performed frequent follow-up assessments while waiting for results.However,after 1 mo,the patient presented with aggressive worsening of the clinical manifestations.Her heart function worsened to NYHA class IV.The laboratory test demonstrated microcytic anemia with increasing brain natriuretic peptide as high as 3939.23 pg/mL(nv < 100) and total homocysteine as high as 119.99 μmol/L(nv < 15).Echocardiography revealed that the pulmonary artery pressure was elevated to 70 mmHg,with a right ventricular Tei index of 0.8,and her pulmonary artery size increased to 30 mm.According to the screening results,a diagnosis of methylmalonic acid was reached.Vitamin B12(1 mg/d) and betaine(200 mg/kg/day) were immediately administered as supplemental therapy with bosentan.After 6 mo of treatment with vitamin B12 and betaine,her pulmonary artery hypertension decreased to 45 mmHg,right ventricular Tei-index dropped to 0.29,and pulmonary artery size decreased to 25 mm,according to the latest echocardiography(Figure 1).Her heart function reversed to NYHA class II.The metabolic disorder analysis failed to identify abnormalities,and a methylmalonic acid level of 6.53 μg/L was observed.
DISCUSSION
Methylmalonic acidemia is an autosomal recessive metabolic disorder that disrupts normal amino acid metabolism.However,this kind of disease rarely demonstrates severe PH as the first clinical manifestation.Defective synthesis of the coenzymes adenosylcobalamin and methylcobalamin has been reported to be the main mediator of PH as a result of mitochondrial dysfunction[4].These disorders might lead to capillary thrombosis in the lung,which would induce PH.In addition,a series of genome sequencing studies that focused on methylmalonic acidemia confirmed that the cobalamin C type was the most common type that would cause aggressive PH[5,6].
According to the European Society of Cardiology and American Heart Association guidelines for the diagnosis and treatment of pediatric PH[1,7],metabolic disorders have been listed in the 5thdivision.However,both of the guidelines only report glycogen storage disease,Gaucher disease and thyroid disorders.Althoughmethylmalonic acidemia was failed to be mentioned,several studies also reported limited cases of methylmalonic acidemia inducing PH,which was first described by Iodiceet al[8].Becket al[9]reviewed a cohort of 36 methylmalonic acidemia patients with cobalamin C defects and found that 7 of them presented PH.
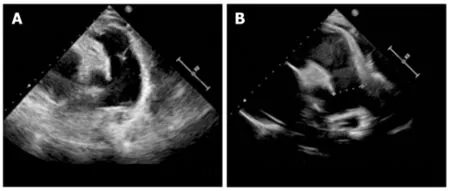
Figure 1 Echocardiography showed the changes in pulmonary artery diameter size before and after treatment.
Electrocardiogram,X-ray,echocardiography,computed tomography,magnetic resonance imaging,pulmonary function test,and catheterization should always be performed as routinely recommended by guidelines.In this case,during the patient's first stay in the hospital,we followed the recommended process for obtaining PH diagnosis.However,we failed to confirm the diagnosis quickly,which aggravated the patient's condition.Then,during the second hospital stay,the combination of genome sequencing and metabolic disorder screening identified methylmalonic acidemia,cobalamin C type.After receiving targeted therapies,this patient's impaired pulmonary artery pressure and heart function were reversed.
CONCLUSION
In summary,as metabolic disorders have already been mentioned in the guidelines,emerging types of metabolic dysfunction have been proven to have a manifestation of PH.It is recommended that genome sequencing and metabolic disorder screening are initially performed to obtain a diagnosis for unexplained or aggressive PH.Based on the literature review and our experience,once the patients presented with the abovementioned symptoms,genome sequencing and metabolic disorder screening should be prioritized rather than performed last to explore possible reasons.We summarized the criteria that indicate a need for these tests,as follows:(1) Failed to detect a structural malformation of the cardiovascular and pulmonary system;(2)Negative results for autoimmune disease;and(3) Aggressive pulmonary artery pressure elevation for a limited time or very early-onset PH without other disorders affecting other systems.
 World Journal of Clinical Cases2020年5期
World Journal of Clinical Cases2020年5期
- World Journal of Clinical Cases的其它文章
- Laparoscopic repair of complete intrathoracic stomach with iron deficiency anemia: A case report
- Growth hormone therapy for children with KBG syndrome: A case report and review of literature
- Hepatoid adenocarcinoma of the stomach: Thirteen case reports and review of literature
- Cerebral venous sinus thrombosis following transsphenoidal surgery for craniopharyngioma: A case report
- Microscopic removal of type lll dens invaginatus and preparation of apical barrier with mineral trioxide aggregate in a maxillary lateral incisor: A case report and review of literature
- Hyoid-complex elevation and stimulation technique restores swallowing function in patients with lateral medullary syndrome:Two case reports