草果總黃酮的大孔吸附樹脂純化工藝優化研究
2020-04-20 01:35:04趙雨鴻沈華代雙億蒲忠慧謝子銳肖靈況敏代敏
中國藥房 2020年7期
趙雨鴻 沈華 代雙億 蒲忠慧 謝子銳 肖靈 況敏 代敏
摘 要 目的:建立草果總黃酮的含量測定方法,并對其大孔吸附樹脂純化工藝進行優化。方法:采用高效液相色譜法測定草果中總黃酮的含量。色譜柱為Eclipse Plus C18,流動相為乙腈-1%醋酸水溶液(15 ∶ 85,V/V),柱溫為40 ℃,流速為0.8 mL/min,檢測波長為256 nm,進樣量為10 μL。以吸附、解吸性能為考察指標,采用靜態吸附和解吸試驗對6種大孔吸附樹脂進行篩選,采用靜態吸附和解吸動力學試驗考察吸附和解吸時間。在單因素試驗的基礎上,以總黃酮含量(以蘆丁計)為評價指標,以上樣液質量濃度、上樣液pH、乙醇體積分數及洗脫用量為考察因素,采用正交設計優化草果總黃酮的純化工藝并進行驗證試驗。結果:蘆丁檢測質量濃度的線性范圍為0.028~0.281 mg/mL(r=0.999 9);定量限為437.5 ng/mL,檢測限為109.4 ng/mL;精密度、穩定性、重復性試驗的RSD均小于2%,加樣回收率為96.24%~99.75%(RSD<2%,n=6)。HPD450型大孔吸附樹脂對草果總黃酮的靜態吸附和解吸綜合能力最適中,最佳靜態吸附與解吸時間均為12 h。最優純化工藝為上樣質量濃度1.854 4 mg/mL,上樣液pH 7,乙醇體積分數60%,乙醇洗脫用量8倍柱體積。驗證試驗顯示,按最優工藝純化后的草果總黃酮含量由純化前的22.556 7 mg/g上升至57.728 2 mg/g,純化倍數為2.56(n=3)。結論:所建含量測定方法靈敏度高、穩定性好,優化的純化工藝穩定、可行,可用于草果總黃酮的純化。
關鍵詞 草果;總黃酮;含量測定;大孔吸附樹脂;純化工藝
ABSTRACT? ?OBJECTIVE: To establish a method for the content determination of total flavonoids from Amomum tsao-ko, and to optimize the purification technology by macroporous resin. METHODS: The content of total flavonoids was measured by HPLC. The determination was performed on Eclipse Plus C18 column with mobile phase consisted of acetonitrile-1% acetic acid solution (15 ∶ 85, V/V) at the flow rate of 0.8 mL/min. The column temperature was 40 ℃, and the detection wavelength was set at 256 nm. The sample size was 10 μL. Taking the adsorption and desorption performance as indexes, 6 kinds of macroporous resins were screened out by static adsorption and desorption tests; adsorption and desorption time were investigated by static adsorption and desorption kinetics tests. Using the content of total flavonoids (calculated by rutin) as index, with sample concentration, sample pH, ethanol volume fraction and elution amount as factors, based on single factor test, orthogonal design was used to optimize the purification technology of total flavonoids from A. tsao-ko, and validation test was performed. RESULTS: The linear range of rutin were 0.028-0.281 mg/mL(r=0.999 9). The limit of quantification was 437.5 ng/mL and the limit of detection was 109.4 ng/mL. RSDs of precision, stability and reproducibility tests were all lower than 2%; the recoveries were 96.24%-99.75%(RSD<2%,n=6). The comprehensive capacity of adsorption and desorption of HPD450 macroporous resin was the most suitable, and the best static adsorption and desorption time both were 12 h. The optimal purification technology was 1.854 4 mg/mL, sample pH value was 7, ethanol volume fraction was 60%, the amount of ethanol elution was 8 times of the column volume. Vertification tests show that after optimized, the content of total flavonoids from A. tsao-ko increased from 22.556 7 mg/g to 57.728 2? ?mg/g. The purity of was 2.56 times higher than before purification. CONCLUSIONS: Established method is sensitive and stable for the content determination. Optimal purification technology is stable and feasible, which is suitable for purifieation of total flavonoids from A. tsao-ko.
KEYWORDS? ?Amomum tsao-ko; Total flavonoids; Content determination; Macroporous absorption resin; Purification technology
草果為姜科豆蔻屬多年生常綠叢生草本植物草果(Amomum tsao-ko Crevost et Lemaire)的干燥成熟果實,主產于我國云南、廣西、貴州等地,越南亦有分布。該藥性溫、味辛,歸脾、胃經,具有燥濕溫中、截瘧除痰的功效[1]。草果作為一種重要的藥食兩用植物,常被作為調味香料用于菜肴烹飪[2];臨床將其用于治療寒濕內阻、脘腹脹痛、痞滿嘔吐、胃功能紊亂、消化不良等癥[1]。現代藥理研究證實,草果具有調節胃腸道功能紊亂、抗氧化、抗菌、抗腫瘤、降脂和降糖等多種藥理作用[3-8]。草果總黃酮作為草果的有效成分群,具有抗氧化、清除自由基的作用[9]。目前,草果總黃酮的提取多采用溶劑提取法,但該法溶劑用量大,所得總黃酮含量不高,還需進一步純化[10]。此外,草果總黃酮的純化主要采用聚酰胺樹脂和大孔吸附樹脂法[11-12]。其中,聚酰胺樹脂純化存在耗時長以及處理、使用過程中容易堵塞等缺點;而大孔吸附樹脂可最大限度地對中藥有效部位去粗取精,具有選擇性好、吸附容量大、再生處理簡便等優點,在黃酮類化合物的分離純化中應用廣泛[13-14]。為此,本研究在現有提取工藝[9]的基礎上,以含量(以蘆丁計)為指標,采用大孔吸附樹脂法純化草果總黃酮,并對其純化工藝進行優化,以期為草果總黃酮提取物的純化和制備提供參考。
1 材料
1.1 儀器
LC-1260型高效液相色譜(HPLC)儀(美國Agilent公司);ME204型分析天平(梅特勒-托利多儀器上海有限公司);BJ-750A型功能粉碎機(德清拜杰電器有限公司);PHS-320型酸度計(成都世紀方舟科技有限公司);THZ-320型臺式恒溫振蕩器(上海精宏實驗設備有限公司)。
1.2 藥材與試劑
草果藥材(批號:20180705)購自四川新綠色藥業科技發展有限公司,經成都中醫藥大學藥學院李敏教授鑒定為姜科豆蔻屬草本植物草果(A. tsao-ko Crevost et Lemaire)的干燥成熟果實。
蘆丁對照品(成都曼斯特生物科技有限公司,批號:MUST-19010210,純度:99.47%);HPD100、HPD300、HPD450、AB-8、X-5、D101型大孔吸附樹脂(中山東鴻化工有限公司,批號分別為20180618、20181126、20180626、20181005、20180716、20190111,粒徑:0.3~1.25 mm);甲醇、乙腈均為色譜純,其余試劑均為市售分析純,水為蒸餾水。
2 方法與結果
2.1 草果總黃酮的提取
稱取草果藥材適量,粉碎,過二號篩。精密稱取一定量的草果粉末,按照文獻報道的提取工藝條件[9]:乙醇體積分數60%、料液比1 ∶ 50(g/mL,下同)、提取溫度60 ℃、超聲(功率:160 W,頻率:60 kHz)提取60 min,趁熱抽濾;重復上述操作2次,合并濾液,濃縮蒸干,即得草果總黃酮粗品(每1 g粗品相當于生藥73.10 g)。
2.2 總黃酮含量測定
采用HPLC法測定草果總黃酮的含量。
2.2.1 蘆丁對照品溶液的制備 精密稱取蘆丁對照品7.02 mg,置于5 mL量瓶中,加適量甲醇超聲(功率:160 W,頻率:60 kHz)使溶解,放冷,再用甲醇定容,搖勻,即得質量濃度為1.404 mg/mL的蘆丁對照品溶液。
2.2.2 供試品溶液的制備 取“2.1”項下所得的草果總黃酮粗品適量,用甲醇溶解,置于5 mL量瓶中,再用甲醇定容,搖勻,即得供試品溶液。
2.2.3 陰性對照溶液的制備 按“2.1”項下方法制得不含草果藥材的陰性樣品,再用適量甲醇溶解并定容于5 mL量瓶中,即得陰性對照溶液。
2.2.4 色譜條件 色譜柱:Eclipse Plus C18(150 mm×3.0 mm,2.7 μm);流動相:乙腈-1%醋酸水溶液(15 ∶ 85,? ? ?V/V);柱溫:40 ℃;流速:0.8 mL/min;檢測波長:256 nm;進樣量:10 μL。
2.2.5 專屬性 取“2.2.1”項下對照品溶液以甲醇稀釋至質量濃度為0.158 mg/mL,另取“2.2.2”“2.2.3”項下供試品溶液和陰性對照溶液各適量,按“2.2.4”項下色譜條件進樣測定,記錄色譜圖。結果,在該色譜條件下,蘆丁的分離度良好,保留時間約為5.95 min,陰性對照對測定無干擾,色譜圖詳見圖1。
2.2.6 標準曲線的繪制 分別精密吸取“2.2.1”項下蘆丁對照品溶液0.1、0.2、0.4、0.6、0.8、1 mL,分別置于不同的5 mL棕色量瓶中,加甲醇定容,搖勻,得質量濃度分別為0.028、0.056、0.112、0.168、0.225、0.281 mg/mL的系列標準溶液,經0.45 μm微孔濾膜濾過后,取續濾液10? ?μL,按“2.2.4”項下色譜條件進樣分析,記錄色譜圖。以蘆丁質量濃度(x,mg/mL)為橫坐標、峰面積(y)為縱坐標進行線性回歸,得回歸方程y=7 468.1x-60.438(r=0.999 9)。結果表明,蘆丁檢測質量濃度的線性范圍為0.028~0.281 mg/mL。
2.2.7 定量限與檢測限 精密吸取“2.2.6”項下質量濃度為0.112 mg/mL的蘆丁對照品標準溶液適量,以甲醇倍比稀釋,按“2.2.4”項下色譜條件進樣分析,分別以信噪比10 ∶ 1、3 ∶ 1計算定量限與檢測限。結果,定量限和檢測限分別為437.5、109.4 ng/mL。
2.2.8 精密度試驗 精密吸取“2.2.6”項下質量濃度為0.112 mg/mL的蘆丁對照品標準溶液適量,按“2.2.4”項下色譜條件連續測定6次,記錄峰面積。結果,蘆丁峰面積的RSD為1.85%(n=6),表明儀器精密度良好。
2.2.9 重復性試驗 取草果總黃酮粗品粉末,共6份,按“2.2.2”項下方法制備供試品溶液,再按“2.2.4”項下色譜條件連續進樣測定6次,記錄峰面積,根據回歸方程計算供試品溶液中總黃酮的質量濃度。結果,供試品溶液中總黃酮的平均質量濃度為0.032 3 mg/mL,RSD為0.60%(n=6),表明本法重復性良好。
2.2.10 穩定性試驗 取同一供試品溶液,分別于室溫下放置0、2、4、6、8、10 h時按“2.2.4”項下色譜條件進樣分析,記錄峰面積。結果,蘆丁峰面積的RSD為0.79%(n=6),表明供試品溶液在室溫放置10 h內穩定。
2.2.11 加樣回收率試驗 取已知含量的草果總黃酮粗品粉末,分別按已知含量的50%、100%、150%加入0.056 mg/mL蘆丁對照品標準溶液,按“2.2.2”項下方法制備低、中、高質量濃度供試品溶液,再按“2.2.4”項下色譜條件進樣分析,記錄峰面積并計算加樣回收率,每質量濃度平行操作6次。結果,低、中、高質量濃度供試品溶液的加樣回收率分別為96.24%、99.75%、98.13%(平均加樣回收率為98.04%),RSD分別為1.83%、1.76%、1.78%(n=6)。
2.2.12 草果總黃酮含量測定 取草果總黃酮粗品或純化后樣品粉末適量,按“2.2.2”項下方法制備供試品溶液,經0.45 μm微孔濾膜濾過后,取續濾液10 μL,再按“2.2.4”項下色譜條件進樣分析,并根據回歸方程計算溶液中總黃酮的質量濃度,并換算為含量(mg/g)。
2.3 大孔吸附樹脂類型的篩選
2.3.1 靜態吸附與解吸試驗 參照文獻方法[15-16]對6種大孔吸附樹脂(HPD100、HPD300、HPD450、AB-8、X-5、D101)進行預處理,并采用靜態吸附與解吸法[17-18]篩選純化草果總黃酮的最佳樹脂類型。
精密稱取6種經預處理的大孔吸附樹脂各1.0 g,平行3份,分別置于50 mL具塞磨口三角瓶中,然后分別加入草果總黃酮水溶液(“2.1”項下所得粗品用水稀釋而得,按“2.2”項下方法測得質量濃度為0.463 6 mg/mL;下同)20 mL,于25 ℃恒溫振蕩器中振搖吸附24 h,充分吸附后,抽濾,將濾液蒸干后用甲醇定容至10 mL,即得吸附液。取上述吸附已達飽和并經洗凈、濾干的樹脂,加入75%乙醇20 mL,于25 ℃恒溫振蕩器中振搖解吸24 h,充分解吸后,抽濾,將濾液蒸干后用甲醇定容至10 mL,即得解吸液。分別吸取上述吸附液和解吸液各1.0 mL,經0.45 μm微孔濾膜濾過后,取續濾液適量,按“2.2.4”項下色譜條件進樣分析,再按“2.2.12”項下方法計算各溶液中總黃酮的含量,并根據公式①~④計算6種大孔吸附樹脂對草果總黃酮的比吸附量、吸附率以及比解吸量、解吸率:
式中,m為樹脂質量(g),c0為上樣液中總黃酮的質量濃度(mg/mL),V0為上樣液的體積(mL),c1為吸附液中總黃酮的質量濃度(mg/mL),V1為吸附液體積(mL),c2為乙醇洗脫液中總黃酮的質量濃度(mg/mL),V2為乙醇洗脫液體積(mL)。靜態吸附和解吸試驗結果見表1。
由表1可見,6種大孔吸附樹脂的吸附率由高到低依次為HPD100>AB-8>HPD300>HPD450>D101>X-5;解吸率依次為HPD450>X-5>AB-8>HPD100>HPD300>D101。盡管HPD100型樹脂的吸附率最高,但解吸率相對較低;而HPD450型樹脂的解吸率最高,且吸附率居中。綜合考慮,選擇HPD450型大孔吸附樹脂作進一步考察。
2.3.2 靜態吸附和解吸動力學考察 精密稱取經預處理的HPD450型大孔吸附樹脂2.0 g,平行3份,分別置于50 mL磨口錐形瓶中,分別加入質量濃度為0.927 2? ? mg/mL的草果總黃酮水溶液40 mL,于25 ℃恒溫振蕩器中振搖,分別于1、2、4、8、12、24 h時吸取上清液4 mL,濃縮蒸干后用甲醇定容至2 mL,即得吸附液。取吸附24 h后的大孔吸附樹脂,用水洗凈并濾干,加入75%乙醇40 mL,于25 ℃的恒溫振蕩器中振搖解吸,分別于1、2、4、8、12、24 h時吸取上清液4 mL,濃縮蒸干后用甲醇定容至2 mL,即得解吸液。分別吸取上述各個時間點的吸附液和解吸液1 mL,經0.45 μm微孔濾膜濾過后,取續濾液適量,按“2.2.4”項下色譜條件進樣分析,按“2.2.12”項下方法計算各溶液中總黃酮的含量,再按“2.3.1”項下方法計算比吸附量和比解吸量。分別以比吸附量和比解吸量(mg/g)為縱坐標、時間(h)為橫坐標,繪制HPD450型大孔吸附樹脂的靜態吸附和解吸動力學曲線,結果詳見圖2。
由圖2A可見,在靜態吸附過程中,HPD450型大孔吸附樹脂的比吸附量在1~12 h內呈上升趨勢,12 h后則吸附飽和;由圖2B可見,在靜態解吸過程中,HPD450型大孔吸附樹脂的比解吸量在1~12 h內波動明顯,而12 h后基本達到平衡。為此,本研究選擇最佳靜態吸附與解吸時間均為12 h。
2.4 草果總黃酮純化工藝優化的單因素試驗
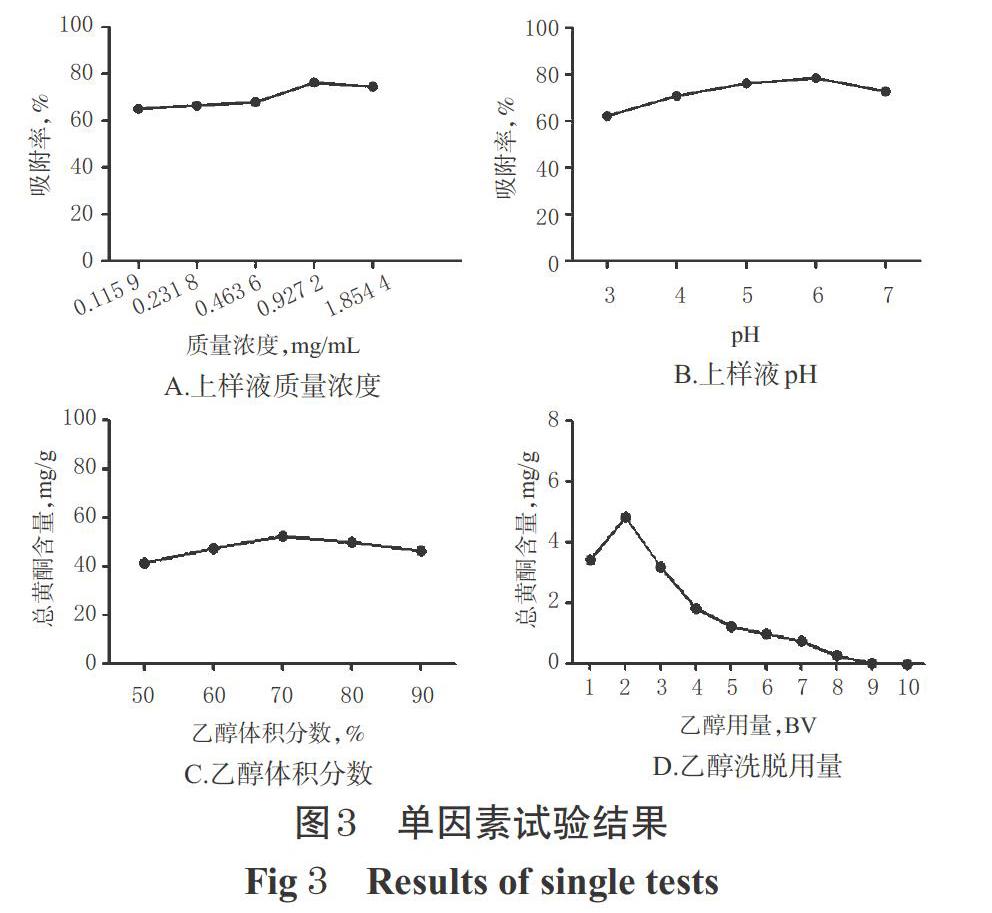
2.4.1 上樣液質量濃度 精密量取5份經預處理的HPD450型大孔吸附樹脂15 mL,濕法裝于徑高比約1 ∶ 13的玻璃柱(直徑:1.5 cm、高:20 cm,下同)中,分別加入pH為6、質量濃度分別為0.115 9、0.231 8、0.463 6、0.927 2、1.854 4 mg/mL的草果總黃酮水溶液20 mL,吸附12 h后,放出吸附液,并用適量水洗脫至流出液無色。將吸附液和水洗脫液合并,濃縮蒸干后,用甲醇定容至2 mL,吸取1 mL,經0.45 μm微孔濾膜濾過后,取續濾液適量,按“2.2.4”項下色譜條件進樣分析,按“2.2.12”項下方法計算各溶液中總黃酮的含量,再按“2.3.1”項下公式②計算吸附率。結果,當質量濃度≤0.927 2 mg/mL時,吸附率有隨上樣液質量濃度增加而上升的趨勢;但當質量濃度>0.927 2 mg/mL時,吸附率不再受上樣液質量濃度增加的影響,反而還略有下降,結果詳見圖3A。綜合考慮,選擇0.927 2 mg/mL為上樣液的最佳質量濃度。
2.4.2 上樣液pH 精密量取5份經預處理的HPD450型大孔吸附樹脂15 mL,置于玻璃柱中,分別加入pH為3、4、5、6、7(pH經1 mol/L鹽酸溶液或1 mol/L氫氧化鈉溶液調節)的0.927 2 mg/mL草果總黃酮水溶液20 mL,吸附12 h后,放出吸附液,并用適量水洗脫至流出液無色。將吸附液和水洗脫液合并,濃縮蒸干后,用甲醇定容至2 mL,吸取1 mL,經0.45 μm微孔濾膜濾過后,取續濾液適量,按“2.2.4”項下色譜條件進樣分析,按“2.2.12”項下方法計算各溶液中總黃酮的含量,再按“2.3.1”項下公式②計算吸附率。結果,當上樣液pH為3~6時,吸附率有隨pH升高而上升的趨勢;但當pH>6時,吸附率反而有所降低,結果詳見圖3B。綜合考慮,選擇pH 6為上樣液的最佳pH。
2.4.3 乙醇體積分數 精密量取5份經預處理的HPD450型大孔吸附樹脂15 mL,置于玻璃柱中,分別加入pH為6的0.927 2 mg/mL草果總黃酮水溶液20 mL,吸附12 h,放出吸附液,并用適量水洗脫至流出液無色后,分別用體積分數為50%、60%、70%、80%、90%的乙醇進行動態解吸。將醇洗脫液濃縮蒸干,用甲醇定容至2 mL,吸取1 mL,經0.45 μm微孔濾膜濾過后,取續濾液適量,按“2.2.4”項下色譜條件進樣分析,按“2.2.12”項下方法計算各溶液中總黃酮的含量。結果,當乙醇體積分數為50%~70%時,大孔吸附樹脂洗脫下來的總黃酮含量有隨乙醇體積分數升高而上升的趨勢;而當乙醇體積分數>70%時,總黃酮含量反而有所降低,這可能與乙醇體積分數越高,其洗脫下來的雜質也越多有關,結果詳見圖3C。綜合考慮,選擇70%為乙醇洗脫的最佳體積分數。
2.4.4 乙醇洗脫用量 精密量取5份經預處理的HPD450型大孔吸附樹脂15 mL,置于玻璃柱中,分別加入pH為6的0.927 2 mg/mL草果總黃酮水溶液20 mL,吸附12 h,放出吸附液,并用適量水洗脫至流出液無色后,再用70%乙醇以1倍柱體積(BV)/h的速度洗脫,分段收集醇洗脫液,每1 BV收集1份,洗脫液濃縮蒸干,用甲醇定容至2 mL,吸取1 mL,經0.45 μm微孔濾膜濾過后,取續濾液適量,按“2.2.4”項下色譜條件進樣分析,按“2.2.12”項下方法計算每BV洗脫液中的總黃酮含量。結果,在第9個BV的洗脫液中幾乎沒有總黃酮,提示此時洗脫基本完成,結果詳見圖3D。綜合考慮,選擇9 BV為最佳乙醇洗脫用量。
2.5 草果總黃酮純化工藝優化的正交試驗
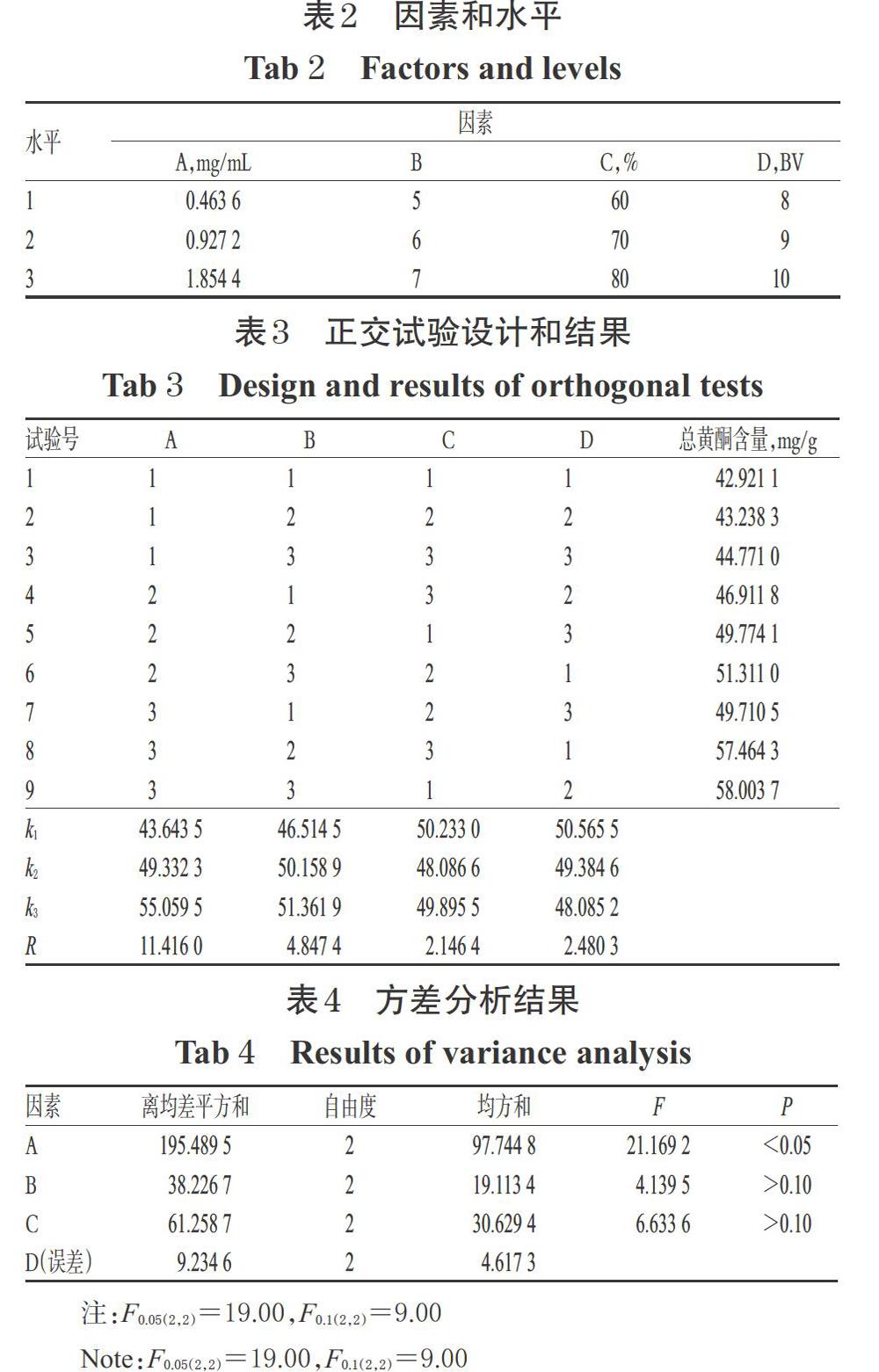
根據單因素試驗結果,以上樣液質量濃度(A)、上樣液pH(B)、乙醇體積分數(C)、乙醇洗脫用量(D)為考察因素,以總黃酮含量為指標,采用L9(34)表設計4因素3水平的正交試驗。因素和水平詳見表2,正交試驗設計和結果詳見表3,方差分析結果見表4。
由表3可見,各因素對總黃酮含量的影響大小依次為A>B>D>C,最優方案為A3B3C1D1。由表4可見,因素A對工藝有顯著影響(P<0.05);B、C對工藝無顯著影響(P>0.05);因素D因離均差平方和最小,故被作為誤差估計項。綜合以上結果,最終確定HPD450型大孔吸附樹脂純化草果總黃酮的最佳工藝條件為A3B3C1D1,即上樣液質量濃度1.854 4 mg/mL,上樣液pH 7,乙醇體積分數60%,乙醇洗脫用量8 BV。
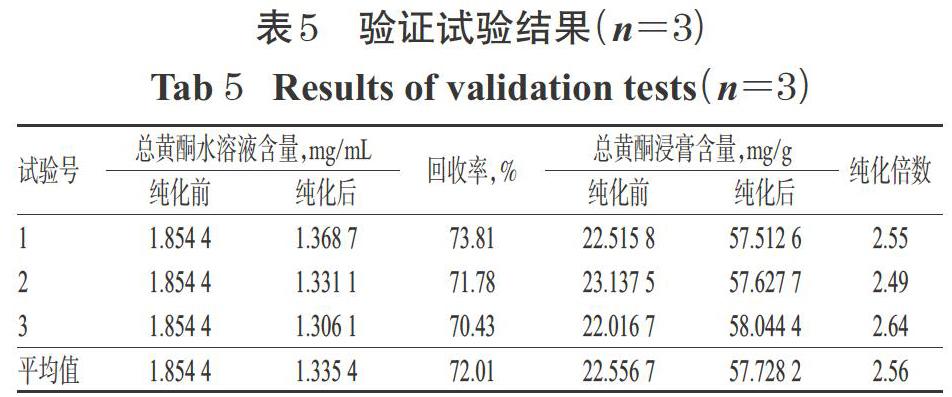
2.6 驗證試驗
按“2.5”項下最佳純化工藝平行操作3次,進行驗證試驗:精密量取經預處理的HPD450型大孔吸附樹脂300 mL,濕法裝柱,加入pH為7、質量濃度為1.854 4? mg/mL的草果總黃酮水溶液400 mL,吸附12 h,放出吸附液,并用適量水洗脫至流出液無色后,再用8 BV 60%乙醇進行洗脫,收集醇洗脫液,濃縮蒸干,用甲醇定容至10 mL,吸取1 mL,經0.45 μm微孔濾膜濾過后,取續濾液適量,按“2.2.4”項下色譜條件進樣分析,再按“2.2.12”項下方法計算總黃酮含量。同時,取純化前后的黃酮水溶液10 mL,置于蒸發皿中蒸干至恒質量,以適量甲醇溶解后,同法測定兩種浸膏中的總黃酮含量。結果,純化后,總黃酮水溶液中的總黃酮平均含量由純化前1.854 4 mg/mL降至1.335 4 mg/mL,平均回收率為72.01%;總黃酮浸膏中的總黃酮平均含量由純化前22.556 7 mg/g升至57.728 2 mg/g,純化倍數為2.56,結果詳見表5。
3 討論
草果是藥食兩用中藥材大宗品種之一,作為香料的用量遠大于藥用,揮發油是其作為香料主要成分之一,2015年版《中國藥典》(一部)也把測定揮發油含量作為評價草果品質的重要指標[1]。為更好地發揮草果藥食兩用的功效,很有必要對其非揮發性成分進行研究。黃酮作為草果中一類重要的非揮發性成分,具有抗氧化、清除自由基的活性[9]。有學者對草果總黃酮進行了提取,但含量不高(24.2 mg/g)[9],且目前尚未見草果總黃酮純化工藝優化的相關報道,故本課題組開展了相關研究。
目前,藥材中黃酮含量的測定方法主要包括紫外分光光度法[19]、HPLC法[20-21]、毛細管電泳法[22]等。其中,HPLC法具有操作簡便、高效、快速的特點,現已被廣泛應用于黃酮的定量分析中[20-21]。黃酮類化合物的分離多采用大孔吸附樹脂和聚酰胺樹脂。與后者相比,大孔吸附樹脂具有吸附容量大、選擇性好、再生簡便、價格低廉等優點,在黃酮類化合物的分離純化中應用廣泛[13-14]。基于此,本研究以草果總黃酮含量為指標,以HPLC法作為定量分析手段,對大孔吸附樹脂的純化工藝進行優化。
本研究選用了非極性(HPD100、HPD300、X-5、D101型)、弱極性(AB-8型)以及極性(HPD450型)大孔吸附樹脂,初步考察了不同類型樹脂對草果總黃酮的富集效果。結果顯示,極性樹脂HPD450型樹脂對草果總黃酮的靜態吸附和解吸綜合能力最適中,與該類成分極性特征對應。本研究在預試驗基礎上,對上樣液質量濃度、pH以及乙醇體積分數、用量進行了單因素試驗,并在單因素試驗的基礎上結合正交試驗對HPD450型大孔吸附樹脂純化草果總黃酮的工藝條件進行了篩選。結果,草果總黃酮純化的最佳工藝條件為上樣液質量濃度1.854 4 mg/mL,上樣液pH 7,水洗除雜后再用8 BV 60%乙醇進行洗脫。值得注意的是,筆者在試驗過程中發現上樣液質量濃度對純化的影響最為顯著,且正交試驗優化所得最佳質量濃度(1.854 4 mg/mL)與單因素篩選質量濃度(0.927 2 mg/mL)相差較大,為避免試驗誤差,筆者曾嘗試將質量濃度升至3.708 8 mg/mL,但發現上樣液中總黃酮呈過飽合,其溶解性大大降低,嚴重影響了純化效果。此外筆者還發現,經50%、90%乙醇洗脫后,草果總黃酮的含量較低;而經60%~80%乙醇洗脫后,總黃酮的含量相對較高,這可能與草果總黃酮極性與60%~80%乙醇更接近有關。驗證試驗結果顯示,純化后,總黃酮水溶液中的總黃酮平均含量由1.854 4 mg/mL降至1.335 4 mg/mL,回收率為72.01%;總黃酮浸膏中的總黃酮平均含量由純化前22.556 7 mg/g升至57.728 2 mg/g,純化倍數為2.56,且3次驗證試驗中總黃酮含量分別為57.512 6、57.627 7、58.044 4 mg/mL,可見優化后的純化工藝穩定,并可有效提高總黃酮的含量。
綜上所述,本研究所建含量測定方法靈敏度高、穩定性好;優化的總黃酮純化工藝穩定、可行。然而,在單因素篩選過程中,草果總黃酮水溶液上樣后,經水洗脫雖可除去大部分水溶性雜質,但本研究并未對水洗脫部位及水洗用量進行考察,可能會造成黃酮類成分的流失。因此,筆者將在后續研究中對此進行探討,并深入研究草果總黃酮的藥理、藥效作用,為草果總黃酮的深入開發和應用提供參考依據。
參考文獻
[ 1 ] 國家藥典委員會.中華人民共和國藥典:一部[S]. 2015年版.北京:中國醫藥科技出版社,2015:239.
[ 2 ] 徐昭璽.百種調料類香料類藥用植物栽培[M].北京:中國農業出版社,2002:243.
[ 3 ] ZHANG TT,LU CL,JIANG JG. Antioxidant and anti-tumour evaluation of compounds identified from fruit of Amomum tsao-ko Crevost et Lemaire[J]. J Funct Foods,2015.DOI:10.1016/j.jff.2015.08.005.
[ 4 ] 張琪,楊揚.草果揮發油對肝癌H22荷瘤小鼠的抑瘤作用[J].武漢大學學報(理學版),2015,61(2):179-182.
[ 5 ] 彭美芳.草果抑菌活性物質作用機制及分離純化的研究[D].海口:海南大學,2014.
[ 6 ] 萬紅焱.滇產草果中有效成分的提取工藝及抗氧化活性研究[D].昆明:昆明理工大學,2015.
[ 7 ] 儲恬予,趙敏吉,于紋婧,等.草果精油對采后草莓保鮮效果及抗氧化活性的研究[J].農產品加工,2019,5(5):24-27.
[ 8 ] ZHANG TT,LU CL,JIANG JG. Bioactivity evaluation of ingredients identified from the fruits of Amomum tsao-ko Crevost et Lemaire,a Chinese spice[J]. Food Funct,2014,5(8):1747-1754.
[ 9 ] 袁園,張瀟,陳碧瓊,等.草果總黃酮的提取及DPPH自由基清除活性研究[J].食品研究與開發,2017,38(15):63-67.
[10] 王婉愉,李姣,張曉峰,等.響應面法優化乙醇提取草果黃酮的工藝研究[J].中國調味品,2018,43(10):164-169.
[11] 同祿祿,李夢耀,許小英,等. HPD-100大孔吸附樹脂與聚酰胺吸附黃酮的對比研究[J]. 應用化工,2016,45(5):929-932、936.
[12] 婁曉晶,李波,陸婷婷,等.大孔樹脂純化鐵皮石斛葉中總黃酮的研究[J].中國現代應用藥學,2019,36(11):1338-1342.
[13] REN J,ZHENG Y,LIN Z,et al. Macroporous resin purification and characterization of flavonoids from Platycladus orientalis(L.)Franco and their effects on macrophage inflammatory response[J]. Food Funct,2017,8(1):86-95.
[14] WAN P,SHENG Z,HAN Q,et al. Enrichment and purification of total flavonoids from Flos Populi extracts with macroporous resins and evaluation of antioxidant activities in vitro[J]. J Chromatogr B Analyt Technol Biomed Life Sci,2014. DOI:10.1016/j.jchromb.2013.11.033.
[15] 蒲忠慧,王力,高宇,等.大孔吸附樹脂純化富集川芎總生物堿的工藝[J].食品研究與開發,2017,38(11):85-91.
[16] 李園園,李洪娟,侯桂革,等.大孔吸附樹脂純化紫菀總黃酮工藝[J].中成藥,2019,41(3):501-505.
[17] 馮宇,劉雪梅,羅偉生,等.大孔樹脂純化荔枝核總黃酮工藝研究[J].中草藥,2019,50(9):2087-2093.
[18] 李俊,劉孟源,方升平,等.大孔吸附樹脂分離純化油橄欖葉總黃酮[J]. 中成藥,2019,41(2):261-265.
[19] 李金洲,陳勇,陳子雋,等. 水石榴中總黃酮的提取工藝優化[J].中國藥房,2019,30(20):2807-2812.
[20] 楊棣華,梁文能.高效液相色譜法測定不同產地蒲黃藥材中3種黃酮苷元含量[J].中國藥業,2019,28(19):27-29.
[21] 孟新濤,魏健,許銘強,等.高效液相色譜法測定不同品種石榴皮中5種類黃酮的含量[J].新疆農業科學,2019,56(9):1659-1667.
[22] 惠陽,仲佳明,武曉雪,等.高良姜黃酮類成分的毛細管電泳特征圖譜分析[J].中藥材,2017,40(1):69-72.
(收稿日期:2019-07-18 修回日期:2020-01-02)
(編輯:張元媛)
