豬苓多糖的PMP柱前衍生化-HPLC指紋圖譜研究
2020-04-20 11:09:08賴戈娜賈文玉羅思婉周昌園黎雄張嫻曾星
中國藥房 2020年7期
賴戈娜 賈文玉 羅思婉 周昌園 黎雄 張嫻 曾星
摘 要 目的:建立豬苓多糖的柱前衍生化-高效液相色譜(HPLC)指紋圖譜,并測定其主要單糖組分含量,為豬苓藥材的質(zhì)量評價提供參考。方法:11批不同產(chǎn)地豬苓藥材經(jīng)水提醇沉、Sevage除蛋白后得到豬苓多糖。多糖經(jīng)三氟乙酸(TFA)水解和1-苯基-3-甲基-5-吡唑啉酮(PMP)衍生化后,進行HPLC分析。色譜柱為HypersiL BDS C18,流動相為0.1 mol/L磷酸鹽緩沖液(pH 6.84)-乙腈(84 ∶ 16,V/V),檢測波長為254 nm,柱溫為30 ℃,流速為1 mL/min,進樣量為20 ?L。采用《中藥色譜指紋圖譜相似度評價系統(tǒng)》(2012A版)建立11批多糖樣品的指紋圖譜并進行相似度評價,通過與對照品比對進行色譜峰指認,再采用SPSS 23.0軟件進行聚類分析,并測定多糖中主要單糖組分的含量。結(jié)果:11批豬苓多糖樣品HPLC指紋圖譜中共呈現(xiàn)出3個共有峰,分別指認為甘露糖、葡萄糖和半乳糖,各批次樣品的相似度均大于0.94。聚類分析將11批多糖樣品分為3類,藥材編號為S1~S6、S8的樣品聚為一類,藥材編號為S7、S10、S11的樣品聚為一類,藥材編號為S9的樣品單獨聚為一類。含量測定結(jié)果顯示,11批樣品中甘露糖含量為1.571~8.771 mg/g、葡萄糖含量為26.072~132.194 mg/g、半乳糖含量為3.420~36.593 mg/g。結(jié)論:本研究建立的柱前衍生化-HPLC指紋圖譜方法可為豬苓的藥材質(zhì)量評價提供參考;不同批次豬苓多糖中單糖組成相同,指紋圖譜特征峰與藥材的產(chǎn)地?zé)o明顯相關(guān)性,藥材之間的單糖含量存在明顯差異。
關(guān)鍵詞 豬苓多糖;單糖;1-苯基-3-甲基-5-吡唑啉酮;柱前衍生化;高效液相色譜法;指紋圖譜
ABSTRACT? ?OBJECTIVE: To establish pre-column derivatization-HPLC fingerprint of Polyporus polysaccharide, and to determine the contents of main monosaccharide components, so as to provide reference for quality evaluation of Polyporus umbellatus. METHODS: Polyporus polysaccharide was extracted with boiling water and precipitated by ethanol and deproteinized by Sevage from 11 batches of P. umbellatus from different producing areas. The samples were firstly hydrolyzed with trifluoro-acetic acid (TFA) and then derivatized by 1-phenyl-3-methyl-5-pyrazolone (PMP). HPLC analysis was then conducted. The determination was carried out on HypersiL BDS C18 column with mobile phase composed of 0.1 mol/L phosphate buffer (pH 6.84)-acetonitrile (84 ∶ 16, V/V) by gradient elution at the flow rate of 1.0 mL/min. The detection wavelength was set at 254 nm, and column temperature was 30 ℃. The sample size was 20 ?L. The similarity of 11 batches of Polyporus polysaccharide was evaluated by using TCM Chromatographic Fingerprint Similarity Evaluation System (2012A edition), and the contents of main monosassharide components were detected. The peak was identified by comparing with the reference substance, and cluster analysis was performed by using SPSS 23.0 software. RESULTS: In HPLC fingerprints of the 11 batches of samples, 3 common peaks were identified, namely mannose, glucose and galactose. The similarity of all samples was above 0.94. Cluster analysis classified 11 batches of samples into three categories. S1-S6, and S8 were grouped into category 1; S7, S10 and S11 were grouped into category 2; S9 was individually grouped into one category. Results of content determination showed that the contents of mannose ranged from 1.571 to 8.771 mg/g; those of glucose ranged from 26.072 to 132.194 mg/g, and those of galactose ranged from 3.420 to 36.593 mg/g. CONCLUSIONS: Established pre-column derivatization HPLC fingerprints can provide reference for quality evaluation of P. umbellatus. The monosaccharide composition of different batches of Polyporus polysaccharide is the same; there is no significant correlation between fingerprint characteristic peak and the origin of herbs; there is significant difference in the content of monosaccharide of P. umbellatus.
KEYWORDS? ?Polyporus polysaccharide; Monosaccharide; PMP; Pre-column derivatization; HPLC; Fingerprint
豬苓又名豬靈芝、野豬苓、豬屎苓、雞屎苓,始載于《神農(nóng)百草經(jīng)》,是多孔菌科真菌豬苓[Polyporus umbellatus(Pers.)Fries]的干燥菌核[1]。其道地藥材產(chǎn)自陜西,此外在河北、河南、云南、四川等地也有分布[2]。2015年版《中國藥典》(一部)豬苓質(zhì)量標(biāo)準(zhǔn)項只收錄了麥角甾醇含量測定[1]。而文獻研究表明,豬苓多糖才是豬苓的主要活性部位,具有調(diào)節(jié)免疫、抗腫瘤、抗炎、抑菌、抗突變、保肝、保腎等與豬苓臨床應(yīng)用相關(guān)的藥理作用[3],且豬苓水提物中多糖含量遠高于麥角甾醇[4],因此應(yīng)將多糖作為中藥豬苓藥材質(zhì)量控制和品質(zhì)評價的指標(biāo)。但目前從豬苓中制備獲得的多糖結(jié)構(gòu)不一,單糖組成各不相同[5-6],豬苓傳統(tǒng)用藥形式水煎液中多糖結(jié)構(gòu)特征不明。多糖一般是由多種比例相對固定的單糖構(gòu)成,通過檢測中藥多糖的單糖組成和摩爾比,可以反映其主要特征,增加鑒別的專屬性,在一定程度上對多糖進行質(zhì)量控制[7-8]。指紋圖譜作為中藥質(zhì)量控制的有效技術(shù)[9-10],目前主要是針對中藥小分子成分,較少用于多糖等生物大分子,這是由大多數(shù)糖類成分無紫外吸收、分子量大、結(jié)構(gòu)復(fù)雜多樣特點決定的[11]。但通過分析各中藥中多糖的主要單糖組成與含量,建立單糖組成指紋圖譜,可以鑒別不同來源的多糖;通過測定其主要組成單糖的絕對含量,可整體評價不同批次藥材質(zhì)量的優(yōu)劣。因此,本研究采用1-苯基-3-甲基-5-吡唑啉酮(PMP)柱前衍生化-高效液相色譜(HPLC)法分析豬苓水煎液多糖的單糖組成和含量,并運用指紋圖譜技術(shù)比較不同產(chǎn)地豬苓多糖中單糖組成與含量差異,為中藥豬苓多糖質(zhì)量標(biāo)準(zhǔn)的建立提供參考。
1 材料
1.1 儀器
1200型HPLC儀(美國Agilent公司);Milli-Q型超純水儀系統(tǒng)(美國Millipore公司);Alpha2-4LSC-Plus型冷凍干燥機(德國Martin Christ公司);RV 10 basic V型旋轉(zhuǎn)蒸發(fā)儀(德國IKA公司);AB135-S型十萬分之一天平、ET58型pH計(德國Mettler-Toledo公司)。
1.2 藥品與試劑
葡萄糖對照品(批號:S10S9I69833,純度:≥98%)、甘露糖對照品(批號:C25D8H51117,純度:≥98%)、半乳糖對照品(批號:Z22J9H64187,純度:≥98%)均購自上海源葉生物科技有限公司;PMP(美國Aladdin公司,批號:C1822095);三氟乙酸(TFA,德國Sigma公司,批號:STBF6942V);乙腈為色譜純,其余試劑均為分析純,水為自制超純水。
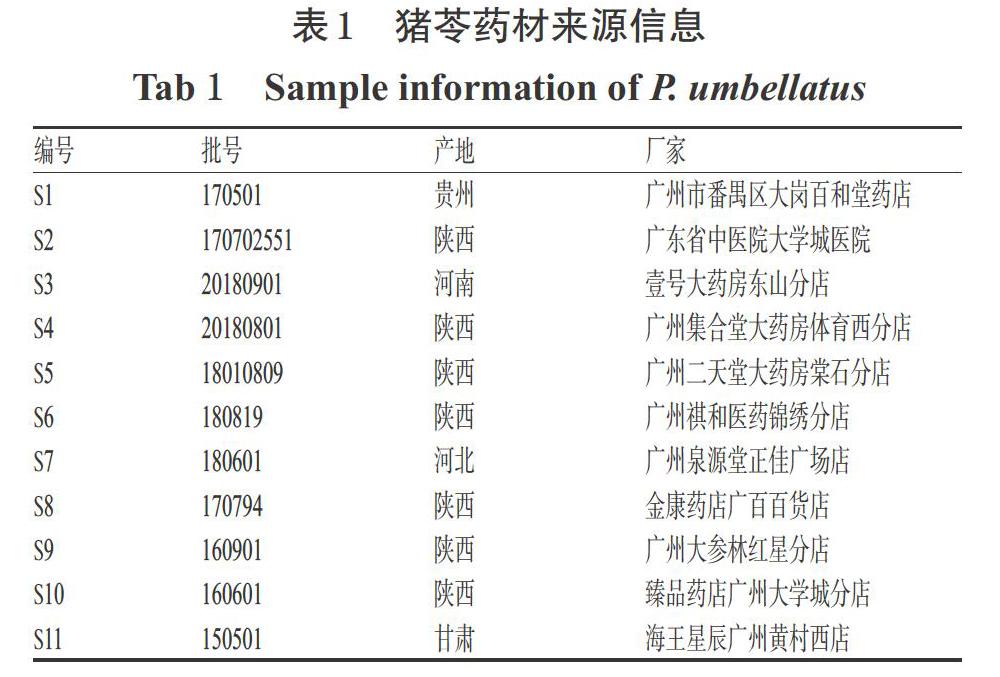
11批豬苓原藥材分別購自不同廠家,經(jīng)廣州中醫(yī)藥大學(xué)第二臨床醫(yī)學(xué)院黎雄副研究員鑒定均為多孔菌科真菌豬苓[P. umbellatus(Pers.)Fries]的干燥菌核,樣品保存于廣州中醫(yī)藥大學(xué)第二臨床醫(yī)學(xué)院Ⅰ期臨床研究分析室。11批豬苓藥材來源信息見表1。
2 方法與結(jié)果
2.1 豬苓多糖的提取和純化
稱取各批次豬苓藥材50 g,粉碎后過60目篩,以料液比為1 ∶ 10(g/mL)的比例加水回流提取2次,每次1.5 h,合并濾液,減壓濃縮至200 mL;加入無水乙醇至乙醇體積分?jǐn)?shù)為80%,靜置過夜;以1 000 r/min離心10 min,沉淀加適量水溶解后,按4 ∶ 1(V/V)的比例加入Sevage試劑[氯仿-正丁醇(4 ∶ 1),V/V],攪拌混勻,去除下層溶劑和中間層蛋白,反復(fù)多次,直至蛋白除盡,冷凍干燥后即得豬苓多糖。采用苯酚-硫酸法[10],以無水葡萄糖為標(biāo)準(zhǔn)品測定11批次豬苓多糖純度,得到各批次藥材樣品(藥材編號:S1~S11)多糖純度分別為43.00%、54.51%、56.05%、52.20%、58.08%、56.60%、60.85%、45.71%、51.19%、50.30%、57.74%。
2.2 溶液的制備
2.2.1 混合對照品溶液的制備 精密稱取甘露糖對照品5.08 mg、葡萄糖對照品4.99 mg、半乳糖對照品5.07 mg,分別用水溶解后定容至5 mL量瓶中,充分搖勻,得各成分單一對照品溶液。分別取各單一對照品溶液1 mL至同一10 mL量瓶中,加水定容至刻度,即得混合對照品溶液。
2.2.2 供試品溶液的制備 精密稱取豬苓多糖20 mg于安瓿瓶中,加入3 mol/L的TFA溶液6 mL溶解,于110 ℃下水解6 h,放冷后,減壓濃縮至干;再加入1 mL甲醇,蒸干,重復(fù)4次以除盡TFA;干燥物加入20 mL水溶解,即得供試品溶液,保存?zhèn)溆谩?/p>
2.2.3 陰性對照溶液的制備 稱取20 mg豬苓多糖(藥材編號:S1)溶于20 mL水中,制備成未經(jīng)水解的陰性對照溶液。
2.3 衍生化處理
分別精密移取“2.2”項下混合對照品溶液、供試品溶液、陰性對照溶液各500 ?L,加入0.3 mol/L的NaOH溶液200 ?L、0.5 mol/L的PMP甲醇溶液200 ?L,充分混勻,在70 ℃水浴中衍生化反應(yīng)60 min;放冷,加入0.3 mol/L的HCl溶液200 ?L中和NaOH,再加入氯仿1 mL充分萃取,重復(fù)萃取3次后,取上層水層液,經(jīng)0.22 ?m濾膜濾過,即得各衍生化溶液。
2.4 色譜條件與系統(tǒng)適用性試驗
色譜柱:HypersiL BDS C18(250 mm×4.6 mm,5 ?m) ;流動相:0.1 mol/L磷酸鹽緩沖液(pH 6.84)-乙腈(84 ∶ 16,V/V);檢測器:紫外檢測器;檢測波長:254 nm;柱溫:30 ℃;流速:1 mL/min;進樣量:20 ?L。取按“2.3”項下方法衍生化處理的混合對照品溶液、供試品溶液(藥材編號:S1)和陰性對照溶液,分別按上述色譜條件進樣分析,記錄色譜圖。結(jié)果,在該色譜條件下,各成分均能達到基線分離,各成分峰間分離度均大于1.5,理論板數(shù)以甘露糖計不低于8 000,且陰性對照溶液不干擾樣品峰的測定。色譜圖詳見圖1。
2.5 HPLC指紋圖譜的建立與分析
2.5.1 精密度試驗 取“2.2.1”項下混合對照品溶液,按“2.3”項下方法進行衍生化后,按“2.4”項下色譜條件連續(xù)進樣6次,記錄峰面積。以葡萄糖峰(峰2)為參照峰,計算得各共有峰相對保留時間的RSD均小于1%(n=6)、相對峰面積的RSD均小于3%(n=6),表明儀器精密度良好。
2.5.2 重復(fù)性試驗 精密稱取同一批號(藥材編號:S1)的豬苓多糖樣品6份,按“2.2.2”項下方法制備供試品溶液后,再按“2.3”項下方法進行衍生化,然后按“2.4”項下色譜條件進樣測定,記錄峰面積。以葡萄糖峰(峰2)為參照峰,計算得各共有峰相對保留時間的RSD均小于1%(n=6)、相對峰面積的RSD均小于3%(n=6),表明該方法重復(fù)性良好。
2.5.3 穩(wěn)定性試驗 稱取豬苓多糖樣品(藥材編號:S1)適量,按“2.2.2”項下方法制備供試品溶液后,再按“2.3”項下方法進行衍生化,然后分別于室溫條件下放置0、2、4、8、10、12 h時按“2.4”項下色譜條件進樣測定,記錄峰面積。以葡萄糖峰(峰2)為參照峰,計算得各共有峰相對保留時間的RSD均小于1%(n=6)、相對峰面積的RSD均小于3%(n=6),表明供試品溶液在室溫下放置12 h內(nèi)穩(wěn)定。
2.5.4 指紋圖譜的生成 取11批豬苓多糖樣品,分別按“2.2.2”項下方法制備供試品溶液后,再按“2.3”項下方法進行衍生化,然后按“2.4”項下色譜條件進樣測定,記錄色譜圖。將11批樣品的色譜圖導(dǎo)入《中藥色譜指紋圖譜相似度評價系統(tǒng)》(2012A版)中,選擇S1號樣品的圖譜作為參照圖譜(S1號圖譜各峰分離完全、信號強度大),設(shè)置時間窗寬度為0.2 min,圖譜間距為40,通過多點校正及自動匹配生成11批衍生化處理的豬苓多糖的疊加指紋圖譜,并采用中位數(shù)法生成對照指紋圖譜。結(jié)果,共得到3個共有色譜峰。11批衍生化處理的豬苓多糖的疊加指紋圖譜詳見圖2,對照指紋圖譜詳見圖3。
2.5.5 相似度評價 以對照指紋圖譜為參照,采用《中藥色譜指紋圖譜相似度評價系統(tǒng)》(2012A版)進行相似度評價。結(jié)果,11批次豬苓多糖的單糖組成的相似度均在0.94以上,表明藥材樣品間差異較小,質(zhì)量穩(wěn)定性良好。11批樣品的相似度評價結(jié)果詳見表2。
2.5.6 色譜峰的指認 通過將HPLC對照指紋圖譜與對照品溶液的HPLC圖譜(見圖1A)比對,指認了峰1為甘露糖、峰2為葡萄糖、峰3為半乳糖。因峰2的分離度良好、峰面積大且穩(wěn)定,故以其保留時間和峰面積為參照,計算得各共有峰相對保留時間的RSD為0~0.29%(n=11),相對峰面積的RSD為0~39.85%(n=11)。結(jié)果表明,不同批次的豬苓多糖雖然具有相同的單糖組成,但是含量差異較大。11批樣品中共有峰的相對保留時間測定結(jié)果詳見表3,相對峰面積測定結(jié)果詳見表4。
2.5.7 聚類分析 將11批豬苓多糖樣品共有峰的相對峰面積導(dǎo)入SPSS 23.0軟件中,以平方歐氏距離作為分類依據(jù),采用Ward法聚類。結(jié)果顯示,當(dāng)歐氏距離為5時,11批豬苓多糖可聚為3類,其中藥材編號為S1~S6、S8的樣品聚為一類,相似度≥0.995;藥材編號為S9的樣品單獨聚為一類,相似度在0.995~0.999之間;藥材編號為S7、S10、S11的樣品聚為一類,相似度在0.946~0.990之間。以上結(jié)果表明,不同來源的豬苓多糖在化學(xué)組成方面具有很好的相似性。11批豬苓總多糖的聚類分析樹狀圖詳見圖4。
2.6 單糖含量的測定
2.6.1 線性關(guān)系考察 精密吸取“2.2.1”項下甘露糖、葡萄糖、半乳糖單一對照品溶液各500 ?L,置于同一5 mL量瓶中,加水定容至刻度,充分混勻。分別吸取上述混合溶液500、250、125、100、62.5、50、25、12.5 ?L至4 mL離心管中,加水稀釋至500 ?L,按“2.3”項下方法進行衍生化,制備系列質(zhì)量濃度的樣品溶液,然后分別按“2.4”項下色譜條件進樣測定,記錄峰面積。以3種單糖的質(zhì)量濃度(x,?g/mL)為橫坐標(biāo)、峰面積(y)為縱坐標(biāo)進行回歸分析。結(jié)果,甘露糖、葡萄糖、半乳糖在各自質(zhì)量濃度范圍內(nèi)線性關(guān)系均良好(r均大于0.999 0),詳見表5。
2.6.2 定量限和檢測限 取“2.2.1”項下混合對照品溶液,加水倍比稀釋,按“2.3”項下方法進行衍生化,然后按“2.4”項下色譜條件進行測定,以信噪比10 ∶ 1、3 ∶ 1分別計算定量限、檢測限。結(jié)果,甘露糖、葡萄糖、半乳糖的定量限分別為0.045 6、0.092 4、0.092 2 μg/mL,檢測限分別為0.022 8、0.030 8、0.026 3 μg/mL。
2.6.3 重復(fù)性試驗 精密稱取同一批樣品(藥材編號:S1)6份,按“2.2.2”項下方法制備供試品溶液,再按“2.3”項下方法進行衍生化,然后按“2.4”項下色譜條件進樣測定,記錄峰面積,并根據(jù)回歸方程計算樣品中3種單糖的含量。結(jié)果,甘露糖、葡萄糖、半乳糖的平均含量分別為3.568、47.332、17.671 mg/g,RSD分別為4.33%、2.77%、3.32%(n=6),表明本方法的重復(fù)性良好。
2.6.4 穩(wěn)定性試驗 取“2.3”項下同一批衍生化處理的供試品溶液(藥材編號:S1),分別于室溫下放置0、2、4、8、10、12 h時按“2.4”項下色譜條件進樣測定,記錄峰面積。結(jié)果,甘露糖、葡萄糖、半乳糖峰面積的RSD分別為4.81%、4.30%、4.49%(n=6),表明該溶液在室溫條件下放置12 h內(nèi)穩(wěn)定性良好。
2.6.5 加樣回收率試驗 取“2.2.2”項下已知含量的供試品溶液(藥材編號:S1)6份,每份1 mL,每份加入15.45 μg/mL的甘露糖、425.68 μg/mL的葡萄糖、95.46? ? μg/mL的半乳糖對照品溶液各1 mL(按“2.2.1”項下方法重新制備),按“2.3”項下方法進行衍生化后,再按“2.4”項下色譜條件進樣測定,記錄峰面積,根據(jù)回歸方程計算各成分的含量并計算其回收率。結(jié)果,甘露糖、葡萄糖、半乳糖的平均加樣回收率分別為99.44%、98.77%、97.57%,RSD分別為2.01%、1.66%、2.03%(n=6),表明該方法準(zhǔn)確度較好,詳見表6。
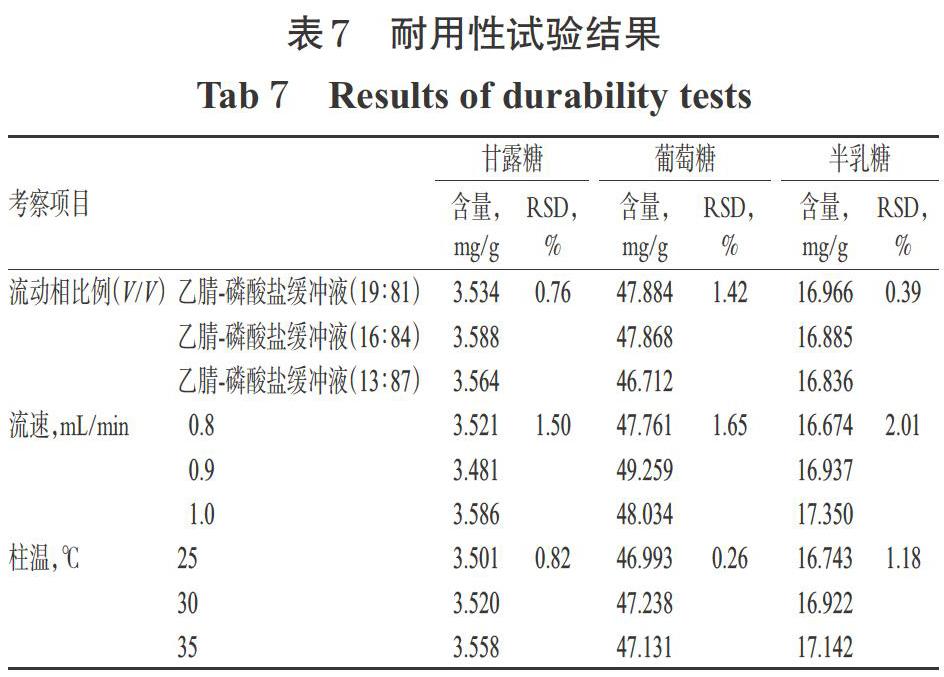
2.6.6 耐用性試驗 取“2.2.2”項下供試品溶液(藥材編號:S1)適量,按“2.3”項下方法衍生化,在其它色譜條件同“2.4”項下不變的情況下,考察不同流動相比例[乙腈-磷酸鹽緩沖液(19 ∶ 81、16 ∶ 84、13 ∶ 87,V/V)]、不同流速(0.8、0.9、1.0 mL/min)、不同柱溫(25、30、35 ℃)對3種單糖含量測定結(jié)果的影響。結(jié)果,3種變化條件下甘露糖含量的RSD分別為0.76%、1.50%、0.82%(n=3),葡萄糖含量的RSD分別為1.42%、1.65%、0.26%(n=3),半乳糖含量的RSD分別為0.39%、2.01%、1.18%(n=3),RSD均小于3%,表明本方法的耐用性較好,詳見表7。
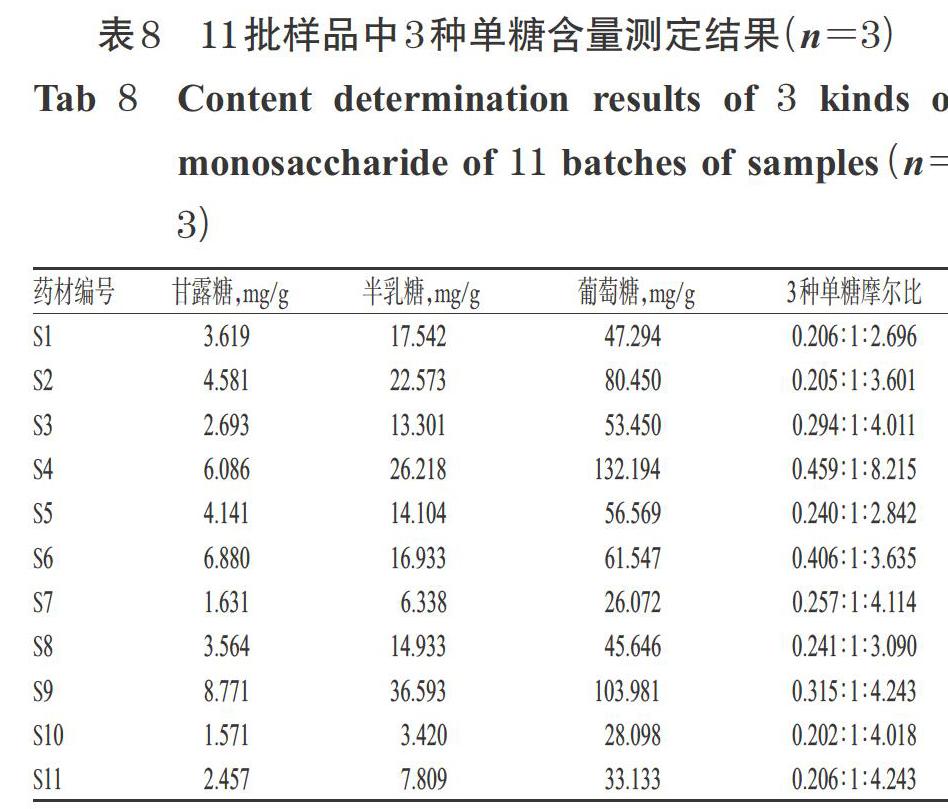
2.6.7 樣品含量測定 按上述所建立的PMP柱前衍生化HPLC法測定11批次豬苓多糖各單糖含量,每個樣品重復(fù)測定3次。結(jié)果,11批樣品中甘露糖含量在1.571~8.771 mg/g之間、半乳糖含量在3.420~36.593之間、葡萄糖含量在26.072~132.194 mg/g之間;不同批次多糖的單糖含量存在明顯差異,但葡萄糖含量均為最高,其次是半乳糖,含量最低的為甘露糖。以半乳糖的摩爾質(zhì)量為基準(zhǔn)1計算各批次多糖的單糖組成摩爾比,得到甘露糖、半乳糖、葡萄糖的摩爾比在(0.202~0.459) ∶ 1 ∶ (2.696~8.215)之間。11批樣品中3種單糖含量測定結(jié)果詳見表8。
3 討論
本課題組前期選擇TFA和PMP的濃度分別為3、0.5 mol/L,以單糖的峰面積為評價指標(biāo),對酸水解的時間以及PMP衍生化的時間和溫度進行了篩選,得到了最佳衍生化條件,即酸水解6 h、70 ℃水浴下衍生化60 min。此外,本課題組前期比較了各單糖在HypersiL BDS C18(250 mm×4.6 mm,5 ?m)、Agilent ZORBAX Eclipse XDB-C18(150 mm×4.6 mm,5 ?m)兩種不同類型色譜柱上的分離效果,結(jié)果發(fā)現(xiàn),長柱比短柱的分離效果好,短柱上葡萄糖峰和半乳糖峰不能完全分離。同時,本課題組前期還考察了0.02、0.05、0.1 mol/L磷酸鹽緩沖液對樣品分離效果的影響,結(jié)果使用0.02、0.05? mol/L的磷酸鹽緩沖液時色譜峰不對稱、拖尾嚴(yán)重;而在0.1 mol/L磷酸鹽緩沖液下樣品色譜峰峰形對稱、分離度好。
本研究以不同產(chǎn)地的11批豬苓藥材為樣品,采用傳統(tǒng)水煎工藝制備總多糖,Sevage法除蛋白后采用PMP柱前衍生化-HPLC法測得其主要單糖組成均為甘露糖、葡萄糖和半乳糖,所測得的豬苓多糖的單糖成分種類較少。分析其原因,可能是未純化的粗多糖中本身含有部分游離單糖,采用Sevage法去除粗多糖中蛋白的同時,也可能除去了游離的單糖和含蛋白比例高的糖肽。本研究基于PMP柱前衍生化-HPLC法建立了11批豬苓多糖的指紋圖譜,采用《中藥色譜指紋圖譜相似度評價系統(tǒng)》(2012A版)計算這11批樣品的相似度后發(fā)現(xiàn),樣品相似度均在0.94以上,這說明不同來源的豬苓多糖樣品在化學(xué)組成方面具有良好的相似性,該制備方法得到的豬苓多糖的質(zhì)量較為穩(wěn)定。聚類分析將11批豬苓多糖分為3類,分類結(jié)果與產(chǎn)地?zé)o相關(guān)性,說明產(chǎn)地因素對多糖的單糖組成影響較小,但這一結(jié)論仍需更多產(chǎn)地、批次的樣品進一步驗證。通過測定豬苓多糖的3種主要單糖含量發(fā)現(xiàn),不同批次豬苓多糖樣品中各單糖含量及單糖摩爾比存在較大差異,但各批次豬苓多糖中單糖含量最高的均為葡萄糖,其次是半乳糖和甘露糖,而單糖的含量差異是否影響其藥理活性,還有待進一步研究。
參考文獻
[ 1 ] 國家藥典委員會.中華人民共和國藥典:一部[S]. 2015年版.北京:中國醫(yī)藥科技出版社,2015:318-319.
[ 2 ] 任潔,李太元,李艷茹,等.長白山豬苓與陜西豬苓的菌絲體轉(zhuǎn)錄組差異比較分析[J].延邊大學(xué)農(nóng)學(xué)學(xué)報,2019,41(3):1-8.
[ 3 ] 夏琴,李敏,周進,等.不同產(chǎn)地、商品規(guī)格及生長年限豬苓麥角甾醇及多糖的含量分析[J].中藥材,2015,38(1):45-48.
[ 4 ] 王天媛,張飛飛,任躍英,等.豬苓化學(xué)成分及藥理作用研究進展[J].上海中醫(yī)藥雜志,2017,51(4):109-112.
[ 5 ] HE P,ZHANG A,ZHANG F,et al. Structure and bioactivity of a polysaccharide containing uronicacid from Polyporus umbellatus sclerotia[J]. Carbohydr Polym,2016.DOI:10.1016/j.carbpol.2016.07.010.
[ 6 ] HE PF,HE L,ZHANG AQ,et al. Structure and chain conformation of an eutral polysaccharide from sclerotia of Polyporus umbellatus[J]. Carbohydr Polym,2017. DOI:10.1016/j.carbpol.2016.08.041.
[ 7 ] 李運,邱國玉,李曉春,等.基于主成分分析的前胡類藥材HPLC指紋圖譜研究[J].藥物分析雜志,2019,39(7):1323-1329.
[ 8 ] 杜澤飛,陶愛恩,夏從龍,等.基于PMP-HPLC和化學(xué)計量學(xué)的黃精基原物種多糖差異分析[J].中國實驗方劑學(xué)雜志,2019,25(15):25-29.
[ 9 ] 鄒純才,鄢海燕.我國中藥色譜指紋圖譜相似度評價方法30年(1988-2017年)研究進展與展望[J].中國中藥雜志,2018,43(10):1969-1977.
[10] 汪海斌,石巖,李芳,等.中藥黃芪指紋圖譜的研究進展[J].中國藥房,2017,28(33):4749-4752.
[11] 李貝貝,盧燕.多糖色譜指紋圖譜的研究方法進展[J].藥學(xué)研究,2018,37(1):41-45.
(收稿日期:2019-11-18 修回日期:2020-01-15)
(編輯:林 靜)
