氧化石墨烯吸附水體中酚類有機污染物的分子動力學模擬
2020-04-14 08:32:44趙超鋒金佳人霍英忠孫陸艾玥潔
無機材料學報 2020年3期
關鍵詞:體系
趙超鋒, 金佳人, 霍英忠, 孫陸, 艾玥潔
氧化石墨烯吸附水體中酚類有機污染物的分子動力學模擬
趙超鋒1, 金佳人1, 霍英忠1, 孫陸2, 艾玥潔1
(1. 華北電力大學 環境科學與工程學院, 資源環境系統優化教育部重點實驗室, 北京 102206; 2. 南開大學 現代光學研究所, 天津 300350)
研究采用分子動力學模擬(Molecular dynamics simulation, MD)的方法, 以苯酚、-萘酚和4-辛基酚為代表, 研究了酚類有機污染物(Phenolic Organic Pollutants, POPs)在氧化石墨烯(Graphene Oxide, GO)上單獨和競爭吸附過程。通過自由能計算得到三種POPs在GO表面的吸附能分別為: 4-辛基酚(41.34 kJ/mol)>-萘酚(33.23 kJ/mol)>苯酚(19.31 kJ/mol)。吸附過程中的主要作用力為POPs的疏水作用, 而分子團簇、范德華相互作用、靜電相互作用以及氫鍵等在一定程度上增加了GO對POPs的吸附能力。在混合體系中, POPs之間存在明顯的競爭吸附現象, 吸附過程包含了直接吸附和形成分子團簇的間接性吸附兩個過程。本研究結果為含POPs水體的治理以及GO材料的設計和篩選提供了一定的理論依據。
酚類有機污染物; 氧化石墨烯; 競爭吸附; 分子動力學模擬
水資源是地球上一切生命活動賴以生存的自然資源, 隨著工業化和城市化的快速發展, 工業廢水和生活污水被肆意排放, 大量的酚類有機污染物(Phenolic Organic Pollutants, POPs)進入自然環境中, 造成嚴重的水體污染[1]。這些POPs進入生態系統之后, 能夠在動植物體內逐漸富集并通過食物鏈的傳遞最終進入人體, 對人體的消化系統、泌尿系統以及神經系統產生毒害作用, 從而嚴重威脅人類的健康[2-4]。因此, 高效去除自然界水體中的POPs是當前亟待解決的環境問題。目前, 對于含POPs水體的處理方法主要有吸附法[5-7]、萃取法[8-9]、膜分離 法[10-11]、生物降解法[12-13]以及催化氧化法[14-15]等。吸附法具有操作簡單、經濟成本低、吸附容量大以及去除效率高等優勢, 在含POPs水體的處置中顯示出良好的去除效果, 因而被廣泛應用于含POPs的污水凈化工藝中。而吸附劑又直接影響到吸附法的應用實效。因此, 篩選出吸附容量大、穩定性強、成本低廉的吸附劑是治理POPs污染水體的重要課題。氧化石墨烯(Graphene Oxide, GO)是一種二維碳基材料, 具有較高的比表面積、優異的化學穩定性以及大量的羥基、羧基等含氧官能團, 并且水溶性良好, 從而引起了廣泛關注[16-18]。研究表明GO對POPs有良好的去除能力[19-20], 例如, 對苯酚的吸附量可達56.56 mg/g[21], 對-萘酚的吸附量高達207.66 mg/g[22]。然而, 其內在吸附機理, 尤其當多種POPs共存時, GO的選擇性吸附以及POPs之間的競爭吸附機制尚不明確。
分子動力學模擬(Molecular dynamics simulation, MD)是一種基于經典牛頓力學方程的分子模擬方法, 被廣泛應用于藥物設計[23]、材料[24]和生物[25]等領域。在環境領域, MD模擬可用于研究目標污染物分子在各種材料界面上的吸附行為[26-27]。大量的研究工作采用MD模擬方法從分子水平上探究吸附過程中材料與目標污染物的相互作用機制, 對不同條件下的實驗結果給予了很好的解釋和預測[5,21,28-29]。由于吸附過程的復雜性, 已有的實驗和理論研究大多針對單一污染物在材料表面的吸附特性, 而在實際應用中往往達不到材料預期的實驗效果。因此, 研究多種污染物共存時的吸附行為對于材料的實際應用更有價值。
本工作以苯酚(phenol)、-萘酚(-naphthol)和4-辛基酚(4-octyl-phenol)為代表, 采用MD模擬方法, 研究POPs在GO表面單獨及競爭吸附過程。觀察POPs在GO表面吸附過程中構型的動態變化, 分析吸附過程中GO與POPs之間的各種相互作用, 結合傘形采樣的方法計算它們的吸附自由能, 從而推斷POPs在GO材料表面吸附的內在作用機理, 為GO材料更廣泛地應用于含POPs水體的治理提供一定理論依據。
1 計算模型與方法
研究采用的GO模型是二維周期性結構, 根據Chen等[30]通過實驗合成的GO材料(O : C的比為0.12), 計算所用的GO模型含有1008個碳原子和126個羥基, O與C的原子比為0.125。其周期性單元結構的尺寸為5.034 nm′4.963 nm, 結構如圖1(a)所示。此外, 苯酚、-萘酚和4-辛基酚的結構如圖1(b)
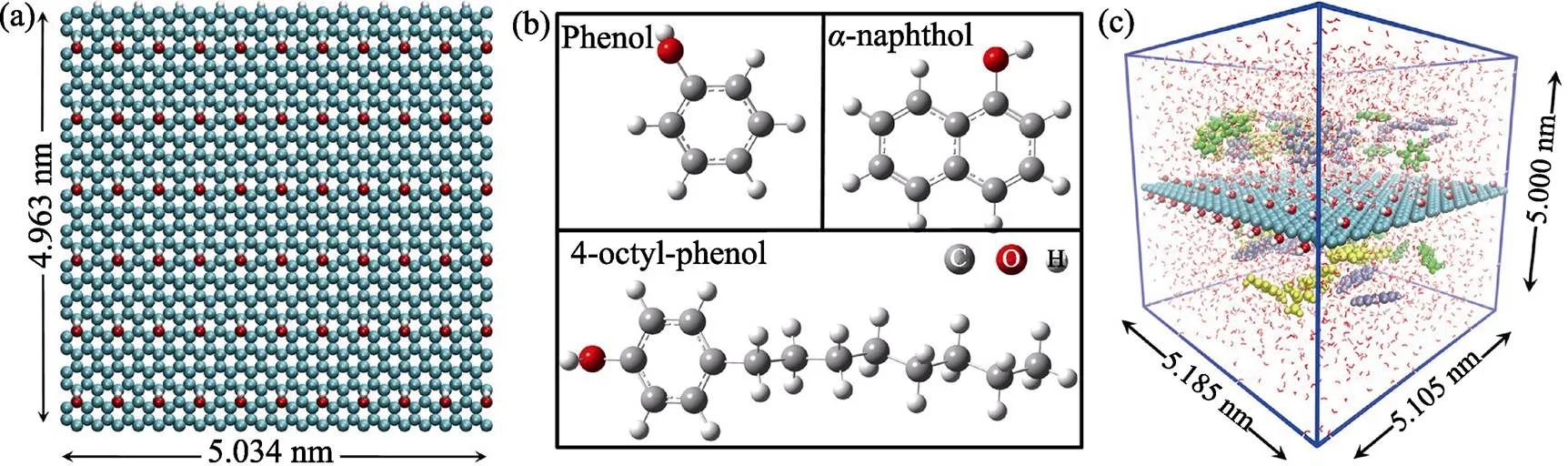
圖1 MD模擬采用的(a)GO模型及其尺寸; (b)苯酚、α-萘酚和4-辛基酚的結構; (c)立方盒子的尺寸及苯酚(綠色)、α-萘酚(紫色)和4-辛基酚(黃色)在GO表面競爭吸附體系的初始結構
所示。從分子結構的角度來說, 三種分子都具有芳香環結構和酚羥基, 與苯酚相比,-萘酚具有兩個芳香環, 而4-辛基酚具有較長的柔性烷基鏈。為了保證GO結構在和方向的周期性, MD模擬采用5.158 nm′5.105 nm′5.000 nm的立方盒子。本研究采用Packmol軟件[31-32]分別在上述立方盒子中構建了GO單獨吸附苯酚(20 個)、-萘酚(20 個)和4-辛基酚(20 個)體系的初始結構(見圖 S1), 以及三種POPs競爭吸附體系(每種分子各20 個)的初始結構(見圖1(c))。模擬體系采用SPC/E水分子模型[33]。OPLS-AA力場是Jorgensen等[34]開發的一種全原子力場, 被廣泛應用于模擬GO和有機化合物的拓撲結構[35-38]。邢寶山課題組[21, 28-29]通過OPLS-AA力場成功模擬了多種芳香族有機污染物在GO表面的吸附過程, 并得到了與實驗觀測相吻合的結果。因此, 本研究采用OPLS-AA力場參數[34]構建了GO和三種POPs的拓撲結構用于動力學模擬, 計算細節詳見補充材料。所有的分子動力學模擬均采用GROMACS 5.0.7軟件[39-40]完成。
2 結果與討論
2.1 苯酚、α-萘酚和4-辛基酚在GO表面的分布
經過100 ns的模擬, 苯酚、-萘酚和4-辛基酚在GO表面單獨吸附的體系均達到平衡, 其穩定的吸附結構如圖2所示。苯酚和-萘酚分子具有獨特的平面芳香環結構, 從圖2(a, b)中可以清晰地看到苯酚和-萘酚分子平行吸附在GO表面。而4-辛基酚分子具有較長的烷基鏈, 從圖2(c)中可以看出它吸附在GO表面的同時還發生分子間聚集, 形成了團簇。另外, 對于三種POPs競爭吸附體系, 從模擬軌跡中截取了吸附過程中幾個典型的吸附結構, 如圖3所示。初始結構中三種POPs均勻地分布在GO兩側。經過10 ns的模擬, 一部分4-辛基酚分子直接吸附在GO表面, 其他的4-辛基酚分子通過與已經吸附在GO表面上的4-辛基酚分子形成較大的團簇吸附到GO表面。與此同時, 溶液中的苯酚和-萘酚分子也逐漸吸附在GO兩側。吸附到25 ns, 溶液中未能直接吸附到GO兩側的苯酚和-萘酚分子通過與4-辛基酚團簇的相互作用間接地吸附在GO表面。從最終時刻(100 ns)的結構圖中可以看出, 溶液中的苯酚、-萘酚和4-辛基酚分子都有效地吸附到GO的兩側。由于吸附位點的限制, 使得某些POPs吸附時不能夠完全平行吸附于GO平面, 而是通過分子間相互作用形成的團簇構成了間接吸附的外層吸附層。
從競爭吸附體系中每一個POPs分子與GO之間的質心距離(見圖4)可以發現以下幾點特征: (1)吸附的類型主要有兩類, 一類是POPs與GO的直接吸附, 吸附距離在0.5 nm以內且波動小, 吸附比較穩定; 另一類是第一吸附層外的間接吸附, 吸附距離在0.5~2.0 nm之間且波動大, 穩定性較差。(2)結合分子動力學的吸附結構以及三種分子的吸附距離可以看出,-萘酚和4-辛基酚相比于苯酚而言, 具有較強的吸附競爭性和穩定性。此外, 分析競爭體系中苯酚、-萘酚、4-辛基酚兩兩之間的相互作用能量(如圖5(a)所示)可以看出, 苯酚和-萘酚在與4-辛基酚團簇相互作用的間接吸附過程中存在明顯的競爭關系。-萘酚和4-辛基酚之間的相互作用逐漸增強, 使得溶液中的-萘酚能夠與GO表面上4-辛基酚團簇緊密結合, 促進了-萘酚的吸附。相反, 苯酚與4-辛基酚之間的相互作用逐漸減弱, 在一定程度上降低了苯酚的吸附效率。
通過分析吸附過程中POPs的聚集程度和徑向分布函數, 進一步探究了POPs在GO表面的分布。在單獨吸附體系中(圖5(b)), 最大的4-辛基酚團簇中分子數的平均值為11.7, 遠大于苯酚(3.6)和-萘酚(8.0), 說明4-辛基酚發生了明顯的聚集。而在競爭吸附體系中(圖5(c)), 苯酚、-萘酚和4-辛基酚最大的團簇中分子數的平均值分別為2.6、4.4和9.2, 均比它們單獨吸附時的小, 表明多種POPs共存時, 不同分子之間的相互影響會降低它們的聚集程度, 從而影響它們的吸附過程。
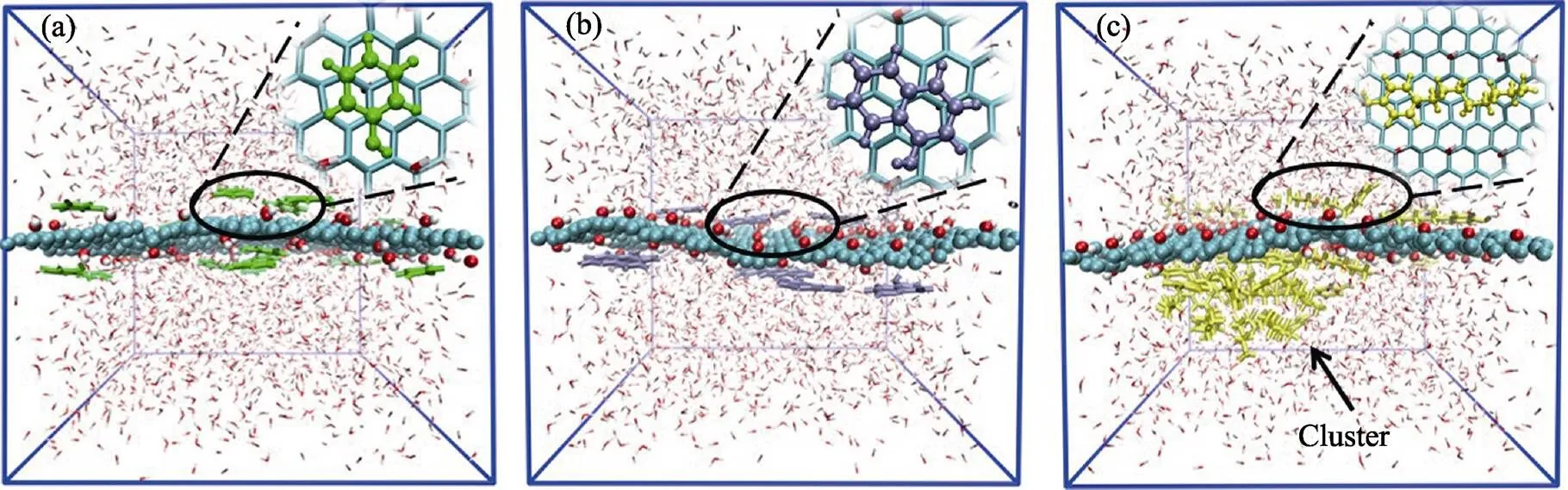
圖2 (a)苯酚、(b)α-萘酚和(c)4-辛基酚在GO表面單獨吸附的平衡結構

圖3 競爭吸附體系中苯酚(綠色)、α-萘酚(紫色)和4-辛基酚(黃色)在GO表面不同時刻的吸附結構圖
Phenol,-naphthol and 4-octyl-phenol molecules are shown as green, purple and yellow molecules, respectively. Water molecules are not shown to highlight the configuration
單獨吸附體系中GO與POPs的徑向分布函數(g())及其積分曲線(n())如圖5(d)所示, 從圖中可以發現, 苯酚、-萘酚和4-辛基酚的g()曲線的整體趨勢是一致的, 都僅有一個強度明顯的峰分布在0.2~0.5 nm范圍內, 說明三種POPs單獨吸附時絕大多數分子都吸附在GO表面的第一吸附層中。此外, 苯酚和-萘酚體系中g()曲線的尖峰比4-辛基酚體系中的強度更大而且更尖銳, 表明吸附在GO表面的苯酚和-萘酚分子的有序度比4-辛基酚分子的高。從n()曲線可以計算出單獨吸附體系中94.8%的苯酚、89.4%的-萘酚、77.2%的4-辛基酚出現在GO的第一吸附層中, 表明GO材料對POPs具有較好的吸附能力, 是一種優良的吸附劑。在競爭吸附體系中(圖5(e)), 三種POPs的g()曲線中尖峰的強度比單獨吸附體系中明顯降低, 說明吸附在GO兩側的POPs的有序度明顯下降, 這與圖3的吸附構型相符合。而且體系中74.9%的苯酚、83.0%的-萘酚、58.3%的4-辛基酚分子出現在GO的第一吸附層中, 與它們單獨吸附時的含量相比均明顯下降, 說明GO表面吸附位點的限制降低了POPs的吸附量。此外, Yu等[5]通過批實驗和光譜分析探究了苯酚和-萘酚在還原氧化石墨烯(rGO)表面的競爭吸附行為。他們的結果表明, 苯酚和-萘酚在吸附過程中存在明顯的競爭關系, 最大吸附量比在單獨吸附體系中均有所下降。理論計算研究揭示了-萘酚由于具有較強的疏水性, 在競爭吸附過程中具有較強的優勢, 說明POPs分子的疏水性在其競爭吸附過程中占主導。這與我們計算的POPs的疏水性強弱順序以及吸附過程主要驅動力的結論相一致。
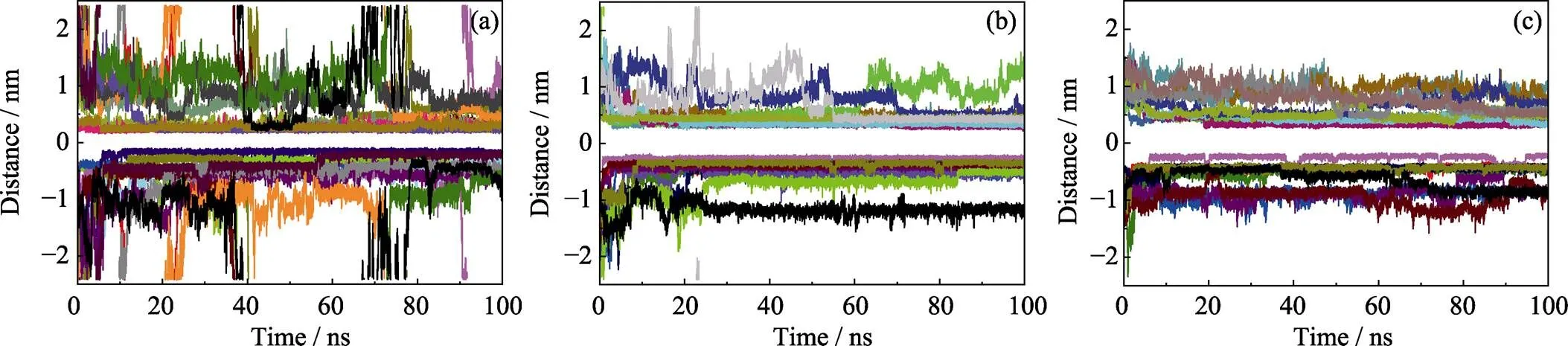
圖4 競爭吸附體系中每一個(a)苯酚、(b)α-萘酚和(c)4-辛基酚分子與GO之間質心距離隨時間的變化
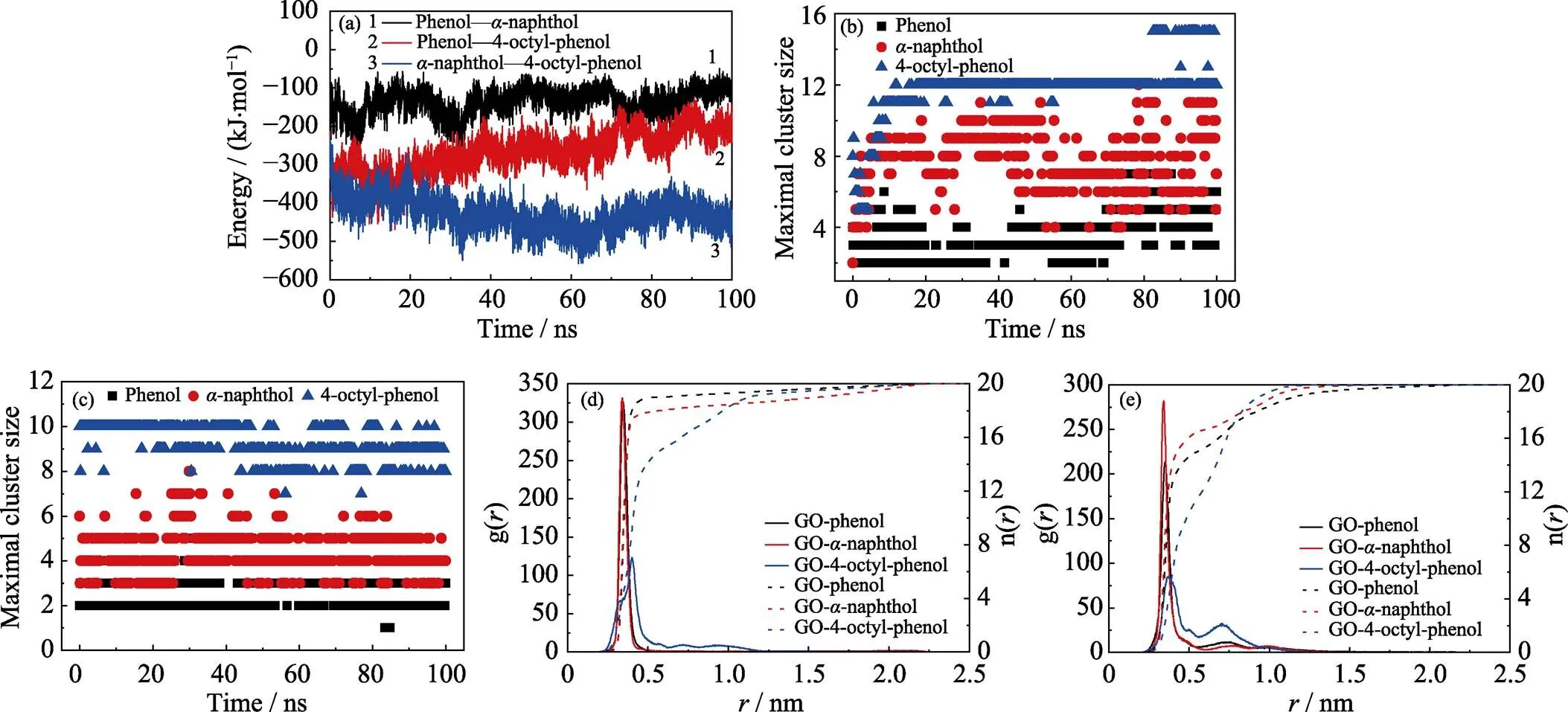
圖5 (a)競爭吸附體系中三種POPs之間的相互作用能量, (b)單獨和(c)競爭體系中POPs最大團簇中的分子數, (d)單獨和(e)競爭體系中POPs在GO表面的徑向分布函數曲線(g(r))以及半徑r范圍內相應的分子數目曲線(n(r))
2.2 GO分別與苯酚、α-萘酚、4-辛基酚的相互作用
通過傘形采樣, 如圖6(a)所示, 計算了苯酚、-萘酚和4-辛基酚在GO表面的吸附能分別為19.31、33.23和41.34 kJ/mol。吸附能的差異與它們自身的分子屬性有很大的關聯。與苯酚相比, 芳香環的增加(-萘酚), 柔性長烷基鏈(4-辛基酚)等都能有效地增強分子與GO的相互作用, 從而提高吸附性能。許多實驗研究發現, GO能夠有效地去除溶液中的POPs分子[30,41]。Zhou等[20]通過改進的Hummers方法結合溶劑熱法以及兩步聚合反應成功制備了 Fe3O4@聚苯胺修飾的磁性氧化石墨烯材料(Fe3O4@PANI-GO), 該材料在水溶液中酚類有機污染物的吸附實驗中表現出良好的吸附容量和優異的磁性分離特性。研究結果表明, 該材料對-萘酚和4-辛基酚的最大吸附量分別為13.19和24.15 mg/g。與-萘酚相比, 4-辛基酚分子的柔性烷基鏈能夠有效地增強它與吸附劑材料的相互作用。該實驗結果與我們模擬的吸附過程中4-辛基酚由于具有較長的柔性烷基鏈使其吸附能大于-萘酚這一理論計算結果相吻合。
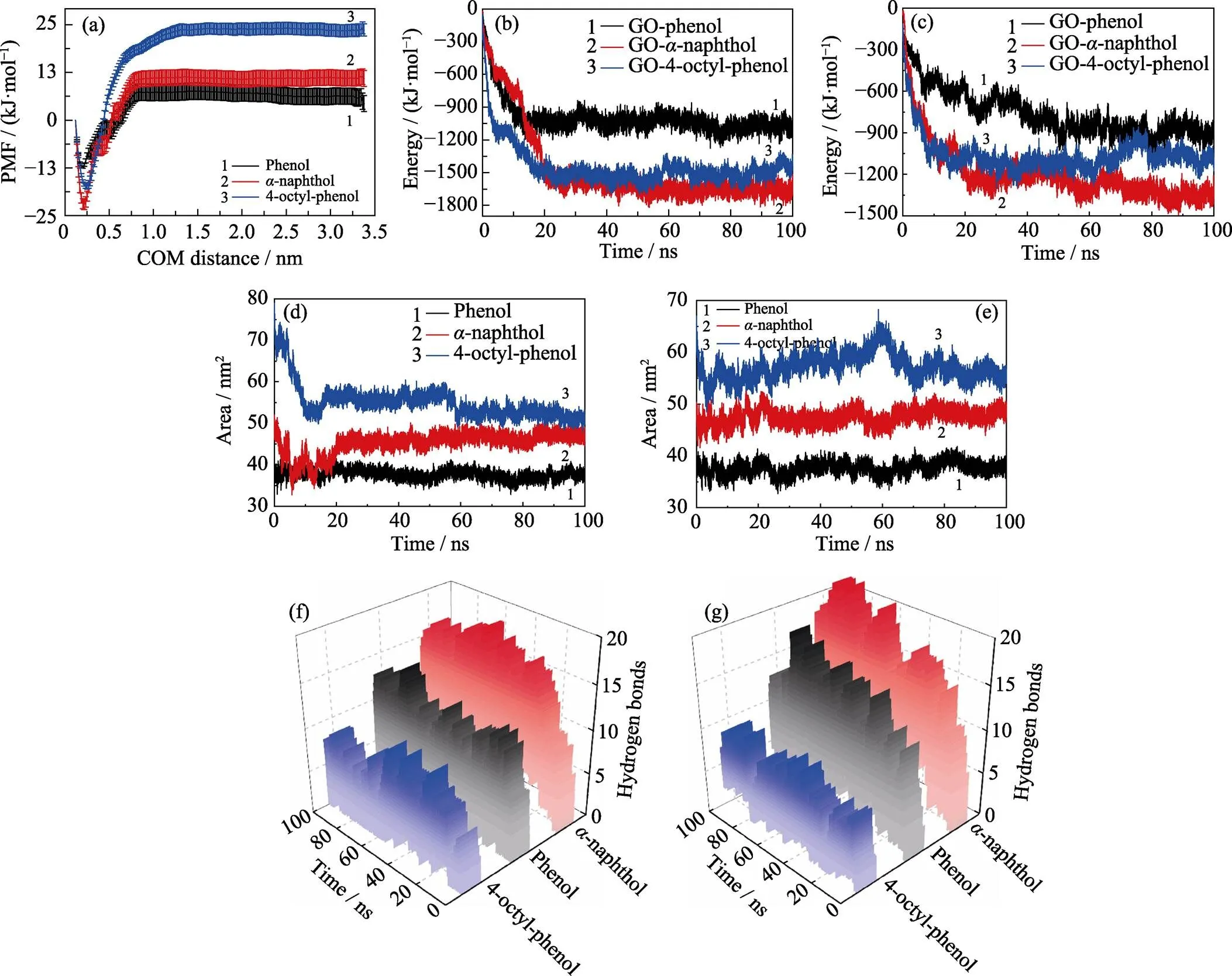
圖6 (a)苯酚、α-萘酚和4-辛基酚分子的吸附自由能; GO與POPs在(b)單獨和(c)競爭體系中的相互作用能量; POPs在(d)單獨和(e)競爭體系中的疏水面積; GO與POPs官能團在(f)單獨和(g)競爭體系中形成的氫鍵數目
圖6(b, c)分別為單獨和競爭吸附體系中GO與POPs之間的相互作用能量, 包括庫侖相互作用能(Coulomb interaction)和蘭納德–瓊斯勢能(Lennard-Jones Potential, L-J Potential)。其中, 庫侖相互作用能的大小可以直接反映出吸附過程中GO與POPs之間靜電相互作用的強弱; 而L-J勢能則是通過計算粒子間的色散力來體現它們之間范德華相互作用的大小。從圖6(b)中可以看出單獨吸附體系中GO與三種POPs之間的相互作用能量的變化趨勢相似: 在前20 ns內迅速下降, 隨后趨于穩定。最終, GO與苯酚、-萘酚、4-辛基酚之間的相互作用能量分別穩定在–1073.91、–1655.69和–1492.28 kJ/mol (取80~100 ns模擬時間范圍內相互作用能量的平均值)。而競爭吸附體系中(見圖6(c)), 在0~60 ns內GO與苯酚之間的相互作用能量緩慢降低, 最終穩定在–891.58 kJ/mol; GO與-萘酚和4-辛基酚的相互作用能量均在前20 ns內迅速降低, 吸附達到平衡時其相互作用能量分別為–1355.32和–1075.89 kJ/mol。將圖6(b, c)中GO與POPs在不同時刻的庫侖相互作用能、L-J勢能以及總的相互作用能量分別匯總在表S1和表S2中, 從表中可以清楚地看出單獨和競爭吸附體系中GO與POPs的L-J勢能分別約占其總能量的80%和75%以上, 均遠大于其庫侖相互作用能, 表明吸附過程中GO與POPs之間的范德華相互作用大于它們之間的靜電相互作用。仔細比較可以發現, 競爭吸附體系中GO與POPs的L-J勢能明顯比它們單獨吸附體系中的小, 而庫侖相互作用能卻有所增加, 這說明多種POPs共存時, 靜電相互作用在吸附過程中的影響增強了。
單獨和競爭吸附過程中POPs團簇的溶劑可及表面積(Solvent Accessible Surface Area, SASA)如圖S2和圖S3所示。從圖中可以看出單獨和競爭吸附過程中三種POPs團簇的疏水面積都遠大于其親水面積。為了更直觀地比較疏水作用對POPs吸附過程的影響, 我們將它們在單獨和競爭吸附過程中疏水面積隨時間變化的曲線匯總在圖6(d, e)中, 從圖中可以發現, 這三種POPs團簇在單獨和競爭吸附體系中疏水作用強弱順序均為: 4-辛基酚>-萘酚>苯酚。分析可知, 三種分子疏水面積的差異主要來源于其分子結構本身, 4-辛基酚分子的長烷基鏈大大增加了它的疏水面積,-萘酚結構中苯環數的增多使其疏水面積大于苯酚。
GO表面的羥基官能團能夠與POPs形成氫鍵。圖6(f, g)分別為單獨和競爭吸附體系中GO與POPs之間形成的氫鍵數目。在單獨(競爭)吸附體系中, GO與苯酚、-萘酚、4-辛基酚分子在模擬的100 ns內形成氫鍵數目的平均值分別為4.9(8.0)、9.7(11.2)和3.7(4.2)。GO與POPs之間形成的氫鍵在一定程度上促進了溶液中POPs在GO表面上的吸附過程, 而且在競爭吸附體系中氫鍵對吸附過程的影響明顯 增強。
3 結論
采用分子動力學模擬的方法, 以苯酚、-萘酚和4-辛基酚為代表, 系統研究了POPs在GO表面的吸附行為。研究結果表明: (1)在單獨吸附的體系中, 溶液中絕大多數的POPs都能夠穩定吸附在GO表面的第一吸附層中。(2)POPs的疏水作用在吸附過程中占主導, 分子團簇、范德華相互作用、靜電相互作用以及氫鍵等在一定程度上也能夠促進POPs在GO表面上的吸附。(3)三種POPs共存時存在明顯的競爭吸附現象, 4-辛基酚由于較強的疏水性和較大的吸附能使其競爭吸附能力最強,-萘酚次之, 苯酚最弱。本研究為研究環境中POPs在GO材料上的吸附行為提供了新的思路和理論基礎。
補充材料:
相關補充材料可以查閱https://doi.org/10.15541/ jim20190377。
[1] AHMARUZZAMAN M. Adsorption of phenolic compounds on low-cost adsorbents: a review., 2008, 143(1/2): 48–67.
[2] BABICH H, DAVIS D L. Phenol: a review of environmental and health risks., 1981, 1(1): 90–109.
[3] KARTHIKEYAN K G, CHOROVER JON, BORTIATYNSKI JACKIE M,Interaction of 1-naphthol and its oxidation products with aluminum hydroxide., 1999, 33(22): 4009–4015.
[4] PONZO OSVALDO J, SILVIA CARBONE. Evidence of reproductive disruption associated with neuroendocrine changes induced by UV-B filters, phthalates and nonylphenol during sexual maturation in rats of both gender., 2013, 311(1/2): 41–51.
[5] YU SHU-JUN, WANG XIANG-XUE, YAO WEN,Macroscopic, spectroscopic, and theoretical investigation for the interaction of phenol and naphthol on reduced graphene oxide., 2017, 51(6): 3278–3286.
[6] YU SHU-JUN, WANG XIANG-XUE, AI YUE-JIE,Experimental and theoretical studies on competitive adsorption of aromatic compounds on reduced graphene oxides., 2016, 4(15): 5654–5662.
[7] PAN BO, LIN DAO-HUI, MASHAYEKHI HAMID,Adsorption and hysteresis of bisphenol a and 17-ethinyl estradiol on carbon nanomaterials., 2008, 42(15): 5480–5485.
[8] RUESGAS-RAMON MARIANA, FIGUEROA-ESPINOZA MARIA CRUZ, DURAND ERWANN. Application of deep eutectic solvents (des) for phenolic compounds extraction: overview, challenges, and opportunities., 2017, 65(18): 3591–3601.
[9] CIULU MARCO, CADIZ-GURREA MARIA DE LA LUZ, SEGURA-CARRETERO ANTONIO. Extraction and analysis of phenolic compounds in rice: a review., 2018, 23(11): 2890–1–20.
[10] CASTRO-MUNOZ ROBERTO, YANEZ-FERNANDEZ JORGE, FILA VLASTIMIL. Phenolic compounds recovered from agro-food by-products using membrane technologies: an overview., 2016, 213: 753–762.
[11] RAZA WASEEM, LEE JECHAN, RAZA NADEEM,Removal of phenolic compounds from industrial waste water based on membrane-based technologies., 2019, 71: 1–18.
[12] ZYSZKA-HABERECHT BEATA, NIEMCZYK EMILIA, LIPOK JACEK. Metabolic relation of cyanobacteria to aromatic compounds., 2019, 103(3): 1167–1178.
[13] SINGH PRIYARAGINI, KUMAR RAKESH. Critical review of microbial degradation of aromatic compounds and exploring potential aspects of furfuryl alcohol degradation., 2019, 27(5): 901–916.
[14] LUO XU-BIAO, DENG FANG, MIN LU-JUAN,Facile one-step synthesis of inorganic-framework molecularly imprinted TiO2/WO3nanocomposite and its molecular recognitive photocatalytic degradation of target contaminant., 2013, 47(13): 7404–7412.
[15] SHAO PENG-HUI, TIAN JIA-YU, YANG FENG,Identification and regulation of active sites on nanodiamonds: establishing a highly efficient catalytic system for oxidation of organic contaminants., 2018, 28(13): 1705295.
[16] BORTHAKUR PRIYAKSHREE, BORUAH PURNA K, DAS MANASH R,Adsorption of 17-ethynyl estradiol and-estradiol on graphene oxide surface: an experimental and computational study., 2018, 269: 160–168.
[17] MOLLA ANIRUDDHA, LI YUAN-YUAN, MANDAL BIKASH,Selective adsorption of organic dyes on graphene oxide: theoretical and experimental analysis., 2019, 464: 170–177.
[18] THAKUR KIRTI, KANDASUBRAMANIAN BALASUBRAMANIAN. Graphene and graphene oxide-based composites for removal of organic pollutants: a review., 2019, 64(3): 833–867.
[19] MUKHERJEE MALOSHREE, GOSWAMI SUDIPTA, BANERJEE PRIYA,Ultrasonic assisted graphene oxide nanosheet for the removal of phenol containing solution., 2019, 13: 398–407.
[20] ZHOU QING-XIANG, WANG YU-QIN, XIAO JUN-PING,Fabrication and characterisation of magnetic graphene oxide incorporated Fe3O4@polyaniline for the removal of bisphenol A,-octyl-phenol, and alpha-naphthol from water., 2017, 7(1): 11316.
[21] TANG HUAN, ZHAO YING, SHAN SU-JIE,Theoretical insight into the adsorption of aromatic compounds on graphene oxide., 2018, 5(10): 2357–2367.
[22] ZHENG HUI-LING, GAO YANG, ZHU KAI-RUO,Investigation of the adsorption mechanisms of Pb(II) and 1-naphthol by beta-cyclodextrin modified graphene oxide nanosheets from aqueous solution., 2018, 530: 154–162.
[23] AMINPOUR MARAL, MONTEMAGNO CARLO, TUSZYNSKI JACK A. An overview of molecular modeling for drug discovery with specific illustrative examples of applications., DOI: 10.3390/molecules24091693
[24] FENG DAI-LI, FENG YAN-HUI, QIU LIN,Review on nanoporous composite phase change materials: fabrication, characterization, enhancement and molecular simulation., 2019, 109: 578–605.
[25] SPONER JIRI, BUSSI GIOVANNI, KREPL MIROSLAV,RNA structural dynamics as captured by molecular simulations: a comprehensive overview., 2018, 118(8): 4177–4338.
[26] MA ZHAO-YANG, PATHEGAMA GAMAGE RANJITH, RATHNAWEERA THARAKA,Review of application of molecular dynamic simulations in geological high-level radioactive waste disposal., 2019, 168: 436–449.
[27] LIU LU-MENG, LIU JUN-JIE, PEI JING-JING. Towards a better understanding of adsorption of indoor air pollutants in porous media— from mechanistic model to molecular simulation., 2018, 11(5): 997–1010.
[28] TANG HUAN, ZHAO YING, YANG XIAO-NAN,Understanding the pH-dependent adsorption of ionizable compounds on graphene oxide using molecular dynamics simulations., 2017, 4(10): 1935–1943.
[29] TANG HUAN, ZHAO YING, SHAN SU-JIE,Wrinkle- and edge-adsorption of aromatic compounds on graphene oxide as revealed by atomic force microscopy, molecular dynamics simulation, and density functional theory., 2018, 52(14): 7689–7697.
[30] CHEN XIAO-XIAO, CHEN BAO-LIANG. Macroscopic and spectroscopic investigations of the adsorption of nitroaromatic compounds on graphene oxide, reduced graphene oxide, and graphene nanosheets., 2015, 49(10): 6181–6189.
[31] MART NEZ JOS MARIO, MART NEZ LEANDRO. Packing optimization for automated generation of complex system's initial configurations for molecular dynamics and docking., 2003, 24(7): 819–825.
[32] MART NEZ LEANDRO, ANDRADE RICARDO, BIRGIN ERNESTO G,PACKMOL: a package for building initial configurations for molecular dynamics simulations., 2009, 30(13): 2157–2164.
[33] BERENDSEN H J C, GRIGERA J R, STRAATSMA T P. The missing term in effective pair potentials., 1987, 91(24): 6269–6271.
[34] JORGENSEN WILLIAM L, MAXWELL DAVID S, TIRADO- RIVES JULIAN. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids., 1996, 118(45): 11225–11236.
[35] WANG JUN-MEI, HOU TING-JUN. Application of molecular dynamics simulations in molecular property prediction I: density and heat of vaporization., 2011, 7(7): 2151–2165.
[36] SUBASINGHEGE DON VISAL, DAVID ROLF, DU PU,Interfacial water at graphene oxide surface: ordered or disordered?, 2019, 123(7): 1636–1649.
[37] PRICE DANIEL J, BROOKS CHARLES L. Detailed considerations for a balanced and broadly applicable force field: a study of substituted benzenes modeled with OPLS-AA., 2005, 26(14): 1529–1541.
[38] KAMINSKI GEORGE A. Accurate prediction of absolute acidity constants in water with a polarizable force field: substituted phenols, methanol, and imidazole., 2005, 109(12): 5884–5890.
[39] BERENDSEN HERMAN J C, VAN DER SPOEL DAVID, VAN DRUNEN RUDI. GROMACS: a message-passing parallel molecular dynamics implementation., 1995, 91(1/2/3): 43–56.
[40] VAN DER SPOEL DAVID, LINDAHL ERIK, HESS BERK,GROMACS: fast, flexible, and free., 2005, 26(16): 1701–1718.
[41] WANG JUN, CHEN BAO-LIANG. Adsorption and coadsorption of organic pollutants and a heavy metal by graphene oxide and reduced graphene materials., 2015, 281: 379–388.
Adsorption of Phenolic Organic Pollutants on Graphene Oxide: Molecular Dynamics Study
ZHAO Chaofeng1, JIN Jiaren1, HUO Yingzhong1, SUN Lu2, AI Yuejie1
(1. MOE Key Laboratory of Resources and Environmental Systems Optimization, College of Environmental Science and Engineering, North China Electric Power University, Beijing 102206, China; 2. Institute of Modern Optics, Nankai University, Tianjin 300350, China)
In this work, molecular dynamics (MD) simulations were applied to address the major concerns about the independent and competitive adsorption processes of phenolic organic pollutants (POPs) on the graphene oxide (GO) in aqueous solution. Phenol,-naphthol and 4-octyl-phenol were adopted as representatives of POPs and their adsorption energies were calculated, which followed an order of 4-octyl-phenol (41.34 kJ/mol)>-naphthol (33.23 kJ/mol)> phenol (19.31 kJ/mol). The simulation results showed that hydrophobic properties of POPs were recognized as the driving force for their adsorption behaviors. Moreover, van der Waals interaction, electrostatic interaction, as well as hydrogen bonds, may also improve the adsorption capacity of GO towards POPs. The competitive adsorption process revealed that in addition to the direct adsorption onto the GO surface, the molecular aggregation may be another indirect adsorption way existed in the mixed system. Understanding the interaction between GO and POPs in aqueous solution is critical to the design and application of graphene-based materials and our findings are believed to contribute further theoretical basis to the engineering treatment of POPs-containing waste water.
phenolic organic pollutants; graphene oxide; competitive adsorption; molecular dynamics simulation
X703
A
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
