超高效液相色譜-四極桿/靜電場軌道阱高分辨質譜法測定豬肉中9 種大環內酯類抗生素
2020-03-11 08:43:34馬俊美曹梅榮范素芳
食品科學 2020年4期
馬俊美,孫 磊,曹梅榮,劉 茁,李 強,*,范素芳,*
(1.河北省食品檢驗研究院,河北 石家莊 050000;2.河北科技大學生物科學與工程學院,河北 石家莊 050018)
大環內酯類抗生素(macrolides antibiotics,MALs)是以大環內酯為母核,與分子糖通過糖苷鍵連接的一類抗生素[1],一般具有12~16 個碳內酯環。MALs易溶于極性有機溶劑,微溶于弱極性溶劑[2-4],對革蘭氏陽性菌及支原體有較強的抗菌活性,同時對部分革蘭氏陰性菌、螺旋體和立克次氏體等也有一定的抗菌活性[5-6],比氨基苷類、多肽類和四環素類等抗生素的毒副作用低,因此被廣泛應用于畜禽疾病的治療[7]。濫用或者使用不當,會導致動物源性食物中MALs的殘留,通過食物鏈進入人體,在體內富集到一定濃度時,會使人眩暈、聽力減退、肝腎受損甚至導致慢性中毒[8-10]。為保障食品安全,日本、美國、歐盟等均制定了動物源性食品中MALs的最高殘留限量,我國農業部也對此做出了規定[11]。為滿足監管的要求,建立一種快速篩查MALs殘留的分析方法是非常必要的。
目前,MALs的檢測方法主要有:微生物法[12]、毛細管電泳-電化學檢測法[13-16]、液相色譜法[17-19]和液相色譜-質譜聯用法[20-28]等,其中,微生物法特異性差、檢出限高,難以準確定性定量[29];液相色譜法是檢測MALs的常用方法,但具有局限性,只能選擇性地檢測某幾種MALs,如紅霉素缺乏特征紫外吸收,較難進行分析;超高效液相色譜-四極桿/靜電場軌道阱高分辨質譜(ultrahigh performance liquid chromatography-quadrupole/orbitrap high resolution mass spectrometry,UPLC-Q-Orbitrap HRMS)將具有高選擇性的四極桿與高分辨率、高靈敏度的軌道阱有機結合,能進行目標物與非目標物的快速篩查與結果確證[30],不同于三重四極桿低分辨質譜使用多反應監測模式進行定量分析,無需對目標物逐個優化子離子及相關參數,彌補傳統三重四極桿質譜檢測化合物數量有限、假陽性誤判的不足,在食品檢測行業中已得到廣泛應用。已有研究報道了UPLC-Q-Orbitrap HRMS應用于動物源性食品中多肽類[31]、磺胺類、喹諾酮類[32]、青霉素類[33]抗生素的測定,本實驗將該技術用于豬肉中MALs篩查、鑒定與定量研究。通過一級全掃描模式獲得目標化合物的精確質量數,進行定性與定量,通過二級全掃描模式獲得目標化合物的二級特征圖譜,進一步提高定性的準確性。本研究能夠準確、高效、快速、高通量地同時檢測多種MALs物質,對保障我國食品安全和提高行業檢測水平具有重要意義。
1 材料與方法
1.1 材料與試劑
甲醇、乙腈(均為色譜純) 德國Merck公司;甲酸(色譜純) 美國Sigma公司;超純水(電阻率18.2 MΩ·cm,25 ℃) 美國Millipore公司;9 種MALs標準品克拉霉素、麥迪霉素、交沙霉素、紅霉素、阿奇霉素、泰樂霉素、羅紅霉素、替米考星、克林霉素(純度≥95%) 中國食品藥品檢定研究院。
1.2 儀器與設備
Ultimate 3000UPLC-Q-Orbitrap HRMS儀美國Thermo Fisher公司;氮吹儀 美國Organomation Associates Jnc公司;3K15高速冷凍離心機 美國Sigma公司;P300H超聲波清洗儀 德國Elma公司;Vortex Genius 3旋渦振蕩器 德國IKA公司。
1.3 方法
1.3.1 樣品前處理
將豬肉樣品充分絞碎混勻后,裝入潔凈的容器內,作為試樣。準確稱取試樣5.00 g,置于50 mL聚丙烯離心管中,加入乙腈20 mL,超聲提取10 min,渦旋5 min充分混勻,8 000hg離心3 min,將上清液轉移至另一離心管中,向殘渣中加入10 mL乙腈重復提取一次。合并上清液,加入氯化鈉2 g和正己烷20 mL,渦旋混勻2 min,8 000hg離心3 min,取乙腈層溶液,于40 ℃氮氣吹干,用1 mL 0.1%甲酸溶液-甲醇(1:1,V/V)溶解殘渣,過0.22 μm濾膜后,準備上機測定。
1.3.2 色譜條件
色譜柱:Waters AcquityB E H C18柱(2.1 mmh100 mm,1.7 μm)。流動相A為0.1%甲酸溶液,B為甲醇。梯度洗脫程序:0~1.5 min,10% B;2~9 min,10%~80% B;9~12 min,80% B;12~13 min,80%~10% B;13~15 min,B相保持10%。
1.3.3 質譜條件
質量分析器:四極桿-靜電場軌道阱;電噴霧離子源(正離子);掃描模式:全掃描/數據依賴二級掃描;鞘氣流量35 arb;輔助氣流量10 arb;噴霧電壓3.8 kV;離子源溫度350 ℃;毛細管溫度320 ℃。一級掃描范圍m/z 100~1 500,一級掃描分辨率70 000,自動增益控制做全掃描S進入C形阱中的目標離子數目3h106。二級掃描分辨率17 500,自動增益控制做MS2進入C形阱中的目標離子數目1h105,隔離窗口為m/z4.0,碰撞能量30 eV。9 種MALs質譜信息見表1。
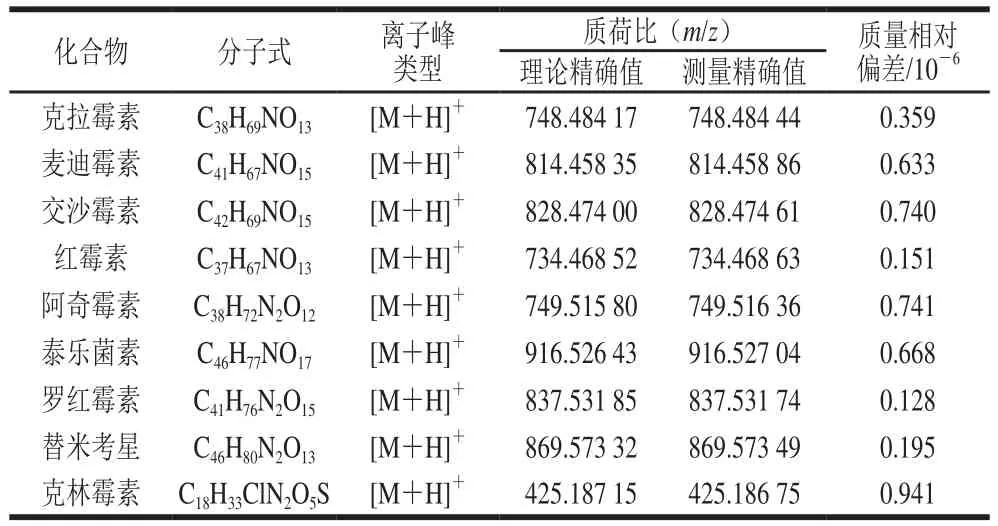
表1 9 種MALs的質譜信息Table 1 Mass spectral parameters for the 9 macrolide antibiotics
2 結果與分析
2.1 提取溶劑的選擇
本實驗選擇6 種不同的溶劑(甲醇、乙腈、0.1%甲酸-乙腈、0.2%甲酸-乙腈、0.1%甲酸-甲醇、0.2%甲酸-甲醇),以9 種MALs的平均回收率(加標量10 μg/kg)考察不同溶劑的提取效果。實驗表明,采用乙腈作為提取溶劑時,9 種MALs的回收率最高。這是因為MALs一般為弱堿性化合物,易溶于有機溶劑,可與酸形成鹽,因此采用中性乙腈提取回收率最高。乙腈提取后,加入正己烷能去除脂肪等雜質,提高回收率,可省去凈化步驟,降低實驗成本。
2.2 色譜-質譜條件的優化
2.2.1 色譜柱的選擇
為得到較好的峰形和分離效果,選擇質量濃度為10 ng/mL的9 種MALs標準品溶液,在相同的流動相和洗脫程序下,分別采用Waters Acquity-BEH C18色譜柱(2.1 mmh100 mm,1.7 μm)、XBridge BEH Shield RP18色譜柱(2.1 mmh100 mm,2.5 μm)、Thermo Accucorea Q C18色譜柱(2.1 mmh100 mm,2.6 μm)、Hypersil Gold C18色譜柱(2.1 mmh100 mm,1.9 μm)進行分離實驗。MALs為堿性化合物,Waters Acquity BEH C18色譜柱采用亞乙基橋雜化顆粒、三鍵鍵合和端基封尾技術,保證了堿性化合物的峰形,相較于其他色譜柱,采用Waters Acquity BEH C18色譜柱可有效分離9 種MALs,且峰形較好。故本實驗選擇Waters Acquity BEH C18(2.1 mmh100 mm,1.7 μm)作為色譜柱。
2.2.2 流動相的選擇
本實驗考察3 種不同的流動相體系(水-甲醇、水-乙腈、0.1%甲酸-甲醇)對色譜峰形和信號響應的影響。實驗表明,使用水-甲醇流動相體系得到的色譜峰形優于水-乙腈體系;使用0.1%甲酸-甲醇作流動相相對于使用水-甲醇體系峰面積變大,穩定性也有所提高。在電噴霧正電離條件下,甲酸的加入有助于目標物離子化,從而提高分離效率和信號的強度。后續實驗選用0.1%甲酸溶液-甲醇體系作為流動相。
2.2.3 質譜條件優化
利用UPLC-Q-Orbitrap HRMS對9 種MALs混合標準溶液進行正離子掃描,得到一級全掃描質譜圖,以各化合物的分子離子峰[M+H]+理論精確質量數提取色譜圖,圖1為9 種MALs的提取離子流色譜圖(50 ng/mL)。9 種MALs的精確質量相對偏差小于1.0h10-6(表1),符合高分辨質譜檢測的要求[34]。二級質譜掃描基于數據依賴的碰撞解離模式運行,采集目標物的二級質譜圖進行定性(圖2)。
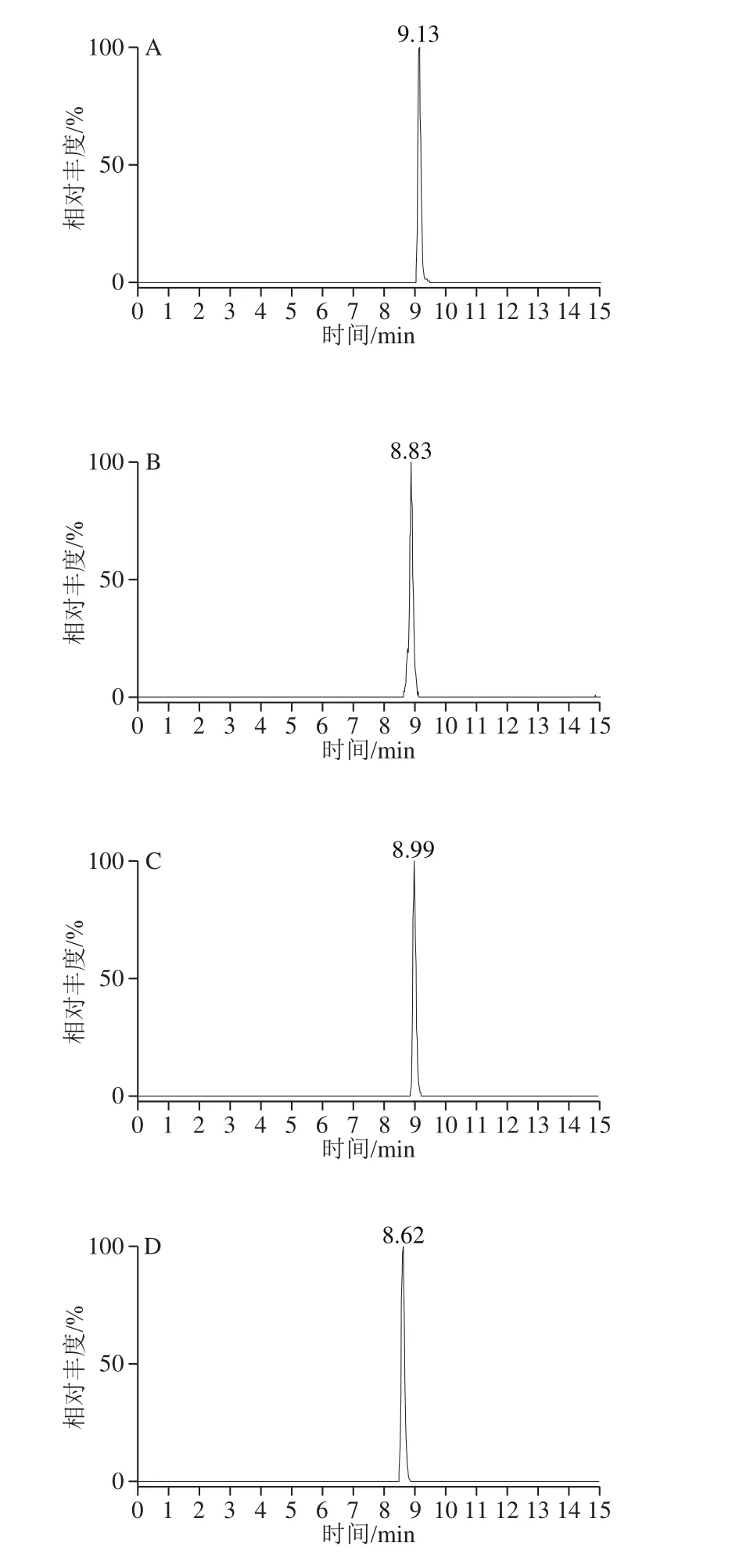
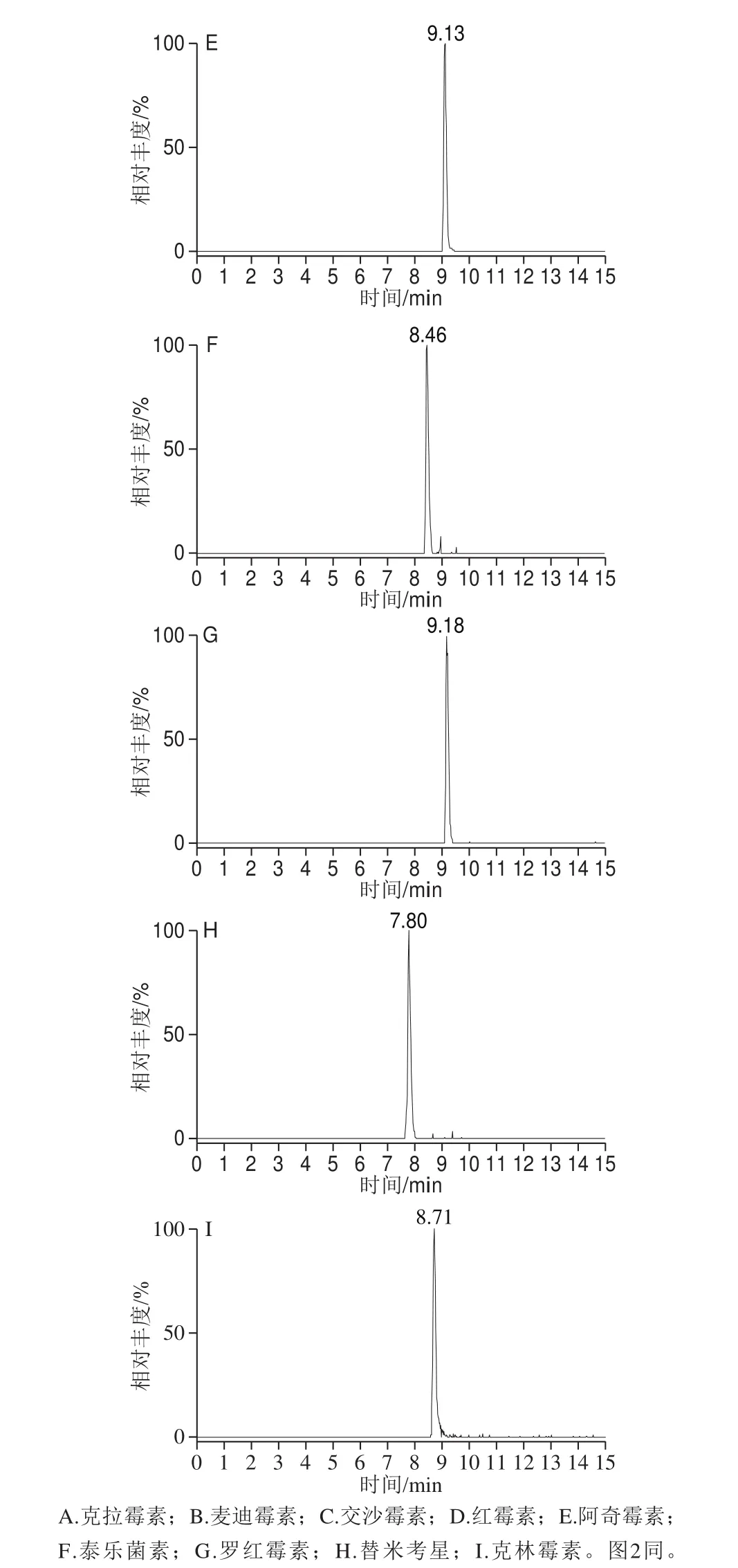
圖1 9 種MALs的提取離子流色譜圖Fig. 1 Extracted ion chromatograms of the 9 macrolide antibiotics
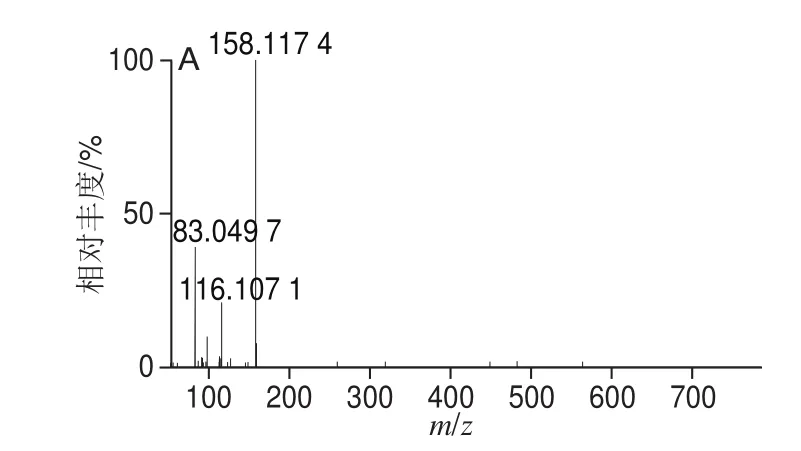
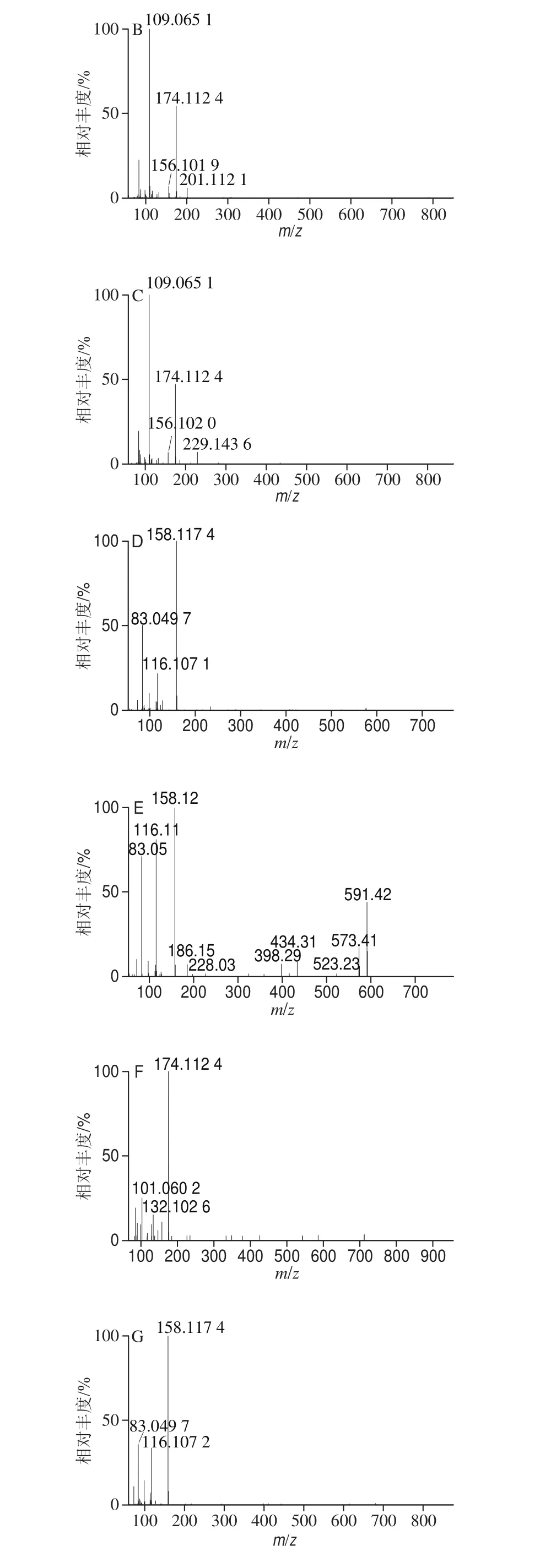

圖2 9 種MALs的二級質譜圖Fig. 2 MS2 spectra of the 9 macrolide antibiotics
2.3 基質效應評價結果
基質是指樣品中目標物以外的組分,與目標物共洗脫出的樣品基質對目標物的離子化過程產生影響,造成離子化抑制或增強,這就是基質效應[35]。與三重四極桿質譜相比,UPLC-Q-Orbitrap HRMS具有更強的抗干擾能力,但基質效應仍然存在,且豬肉樣品基質較為復雜,因此,需要對基質效應進行評價,本實驗將9 種MALs標準品分別用乙腈和豬肉空白基質提取液配制標準溶液(0.5~100 ng/mL),以相同物質在基質溶液中標準曲線斜率與純溶劑中標準曲線斜率比值評價基質效應情況。基質效應大于100%時,表明基質對目標物離子化有增益作用;基質效應小于100%時,表明基質對目標物離子化有抑制作用。結果表明,9 種MALs在豬肉基質中均存在明顯的離子抑制現象:克拉霉素(69.5%)、麥迪霉素(72.9%)、交沙霉素(69.8%)、紅霉素(79.1%)、阿奇霉素(69.4%)、泰樂菌素(71.8%)、羅紅霉素(59.4%)、替米考星(51.9%)、克林霉素(49.2%)。由于基質效應的存在,本方法在定量時采用基質配標,降低基質效應的影響。
2.4 方法的線性范圍、檢出限和定量限結果
在豬肉空白提取液中添加標準溶液配制一系列的標準工作液,以9 種MALs的一級質量數提取離子峰面積為縱坐標(Y),各組分的質量濃度為橫坐標(X)作線性回歸方程,以3 倍和10 倍信噪比對應的空白豬肉樣品添加質量濃度作為檢出限和定量限。9 種MALs的線性方程、檢出限、定量限見表2。結果表明,9 種MALs在0.5~100 μg/L質量濃度范圍內相關系數(r2)大于0.998,線性關系良好。檢出限為0.05~0.2 μg/kg,定量限為0.1~0.4 μg/kg,低于GB/T 20762ü2006《禽畜肉中林可霉素、竹桃霉素、紅霉素、替米考星、泰樂菌素、克林霉素、螺旋霉素、吉他霉素、交沙霉素殘留量的測定液相色譜-串聯質譜法》[36](檢出限為1.0 μg/kg)和SN/T 1777.2ü2007《動物源性食品中大環內酯類抗生素殘留測定方法》[37](測定低限為20 μg/kg)。
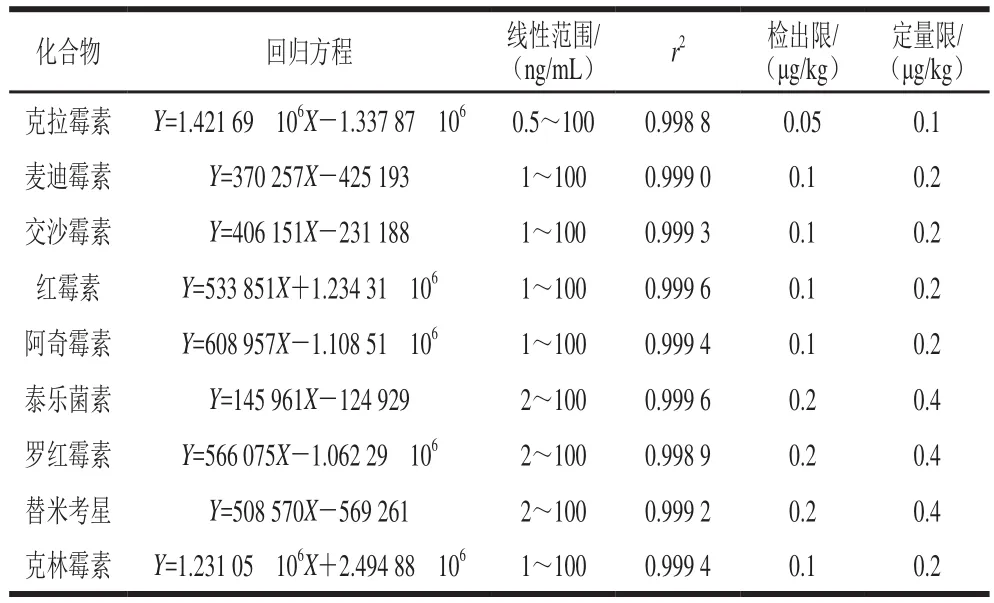
表2 9 種MALs的線性關系、檢出限、定量限Table 2 Linear equations, limits of detection and limits ofquantification of the 9 macrolide antibiotics
2.5 回收率和精密度結果
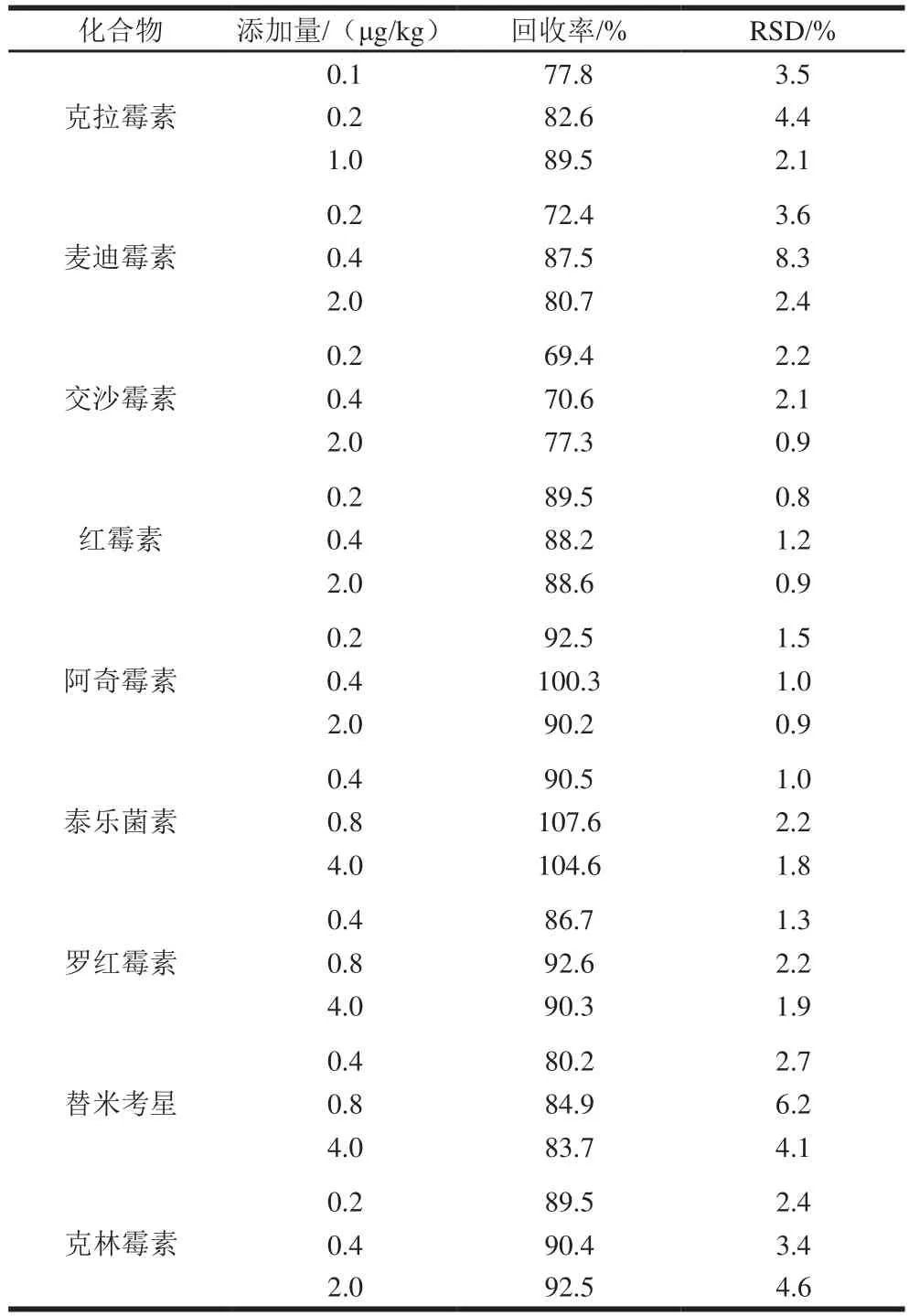
表3 9 種MALs的回收率和精確度Table 3 Spiked RSDs of the 9 macrolide antibiotics
在空白豬肉樣品中添加9 種MALs標準溶液制成陽性樣品,添加水平分別為1、2 倍和10 倍定量限,每個添加水平進行6 次重復實驗,計算回收率和相對標準偏差(relative standard deviations,RSD)。如表3所示,添加量0.1~4.0 μg/kg時,方法的回收率范圍為69.4%~107.6%,RSD不高于8.3%,符合檢驗方法要求。
2.6 實際樣品檢測結果
應用本研究建立的方法對市場采購的20 批次豬肉樣品進行檢測,采用精確質量數和保留時間對樣品中的MALs定性篩查,并結合二級特征碎片離子確證,均未檢出陽性樣品。與標準方法GB/T 20762ü2006[36]和SN/T 1777.2ü2007[37]的測定結果一致。
3 結 論
本研究建立了UPLC-Q-Orbitrap HRMS測定豬肉中9 種MALs,并進行了方法學驗證。該方法簡單高效、選擇性好、靈敏度高,滿足大量豬肉樣品中MALs快速、準確定量的需要,可為其他基質中其他MALs的快速篩查提供方法參考,也是對現有國家標準檢測動物源性食品中MALs的有力補充。
