原位漫反射紅外光譜研究Ni-Fe/γ-Al2O3催化CO甲烷化反應
2020-02-22 00:14:52武瑞芳王永釗趙永祥
工業催化 2020年12期
關鍵詞:催化劑
武瑞芳,王 寧,周 翔,王永釗,趙永祥
(山西大學化學化工學院,精細化學品教育部工程研究中心,山西 太原 030006)
近年來,質子交換膜燃料電池因其環境友好、操作溫度低且工作效率高而受到關注[1-4]。氫氣是質子交換膜燃料電池的燃料,主要通過蒸汽重整和碳氫化合物的部分氧化等途徑制得[5-7],但上述反應制得的氫氣中不可避免地含有體積分數1%~2%的CO,極易毒化燃料電池的電極[8-10]。富氫氣體中微量CO的脫除廣泛采用CO甲烷化法,在報道的眾多CO甲烷化催化劑中,Ni基催化劑因其催化活性高,選擇性好以及成本相對低廉等優勢受到研究者的青睞[11]。
在CO甲烷化反應中,雙金屬Ni基催化劑顯示出比單金屬催化劑更高的催化活性和穩定性[12-17]。本課題組前期研究工作表明,與Ni/γ-Al2O3和Fe/γ-Al2O3催化劑相比,Ni-Fe/γ-Al2O3雙金屬催化劑表現出更優異的CO甲烷化催化性能。在相同反應條件下,220 ℃時Fe/γ-Al2O3催化劑未表現出CO甲烷化催化活性,Ni/γ-Al2O3催化劑上CO轉化率僅為48%,而Ni-Fe/γ-Al2O3雙金屬催化劑上CO則完全甲烷化。通過XRD、N2物理吸附-脫附、H2-TPR、H2-TPD等表征結果顯示,3種催化劑的織構性質并無明顯差異,但Ni-Fe/γ-Al2O3催化劑還原后形成Ni-Fe合金,H2吸附量明顯增加[18]。為了進一步探究Ni/γ-Al2O3、Fe/γ-Al2O3和Ni-Fe/γ-Al2O3催化劑催化活性差異的原因,本文采用原位漫反射紅外光譜對3種催化劑上CO、CO-H2混合氣的吸附行為進行比較研究,旨在闡明催化劑上吸附物種的演變規律,為高性能CO甲烷化催化劑的研發提供理論參考。
1 實驗部分
1.1 催化劑制備
稱取一定量Ni(NO3)2·6H2O和Fe(NO3)2·9H2O配制成單一鹽水溶液和一定計量比的混合鹽水溶液,然后分別等體積浸漬于γ-Al2O3載體上[(40~60)目,比表面積253 m2·g-1],充分攪拌并靜置0.5 h后,120 ℃干燥3 h,空氣氣氛400 ℃焙燒3 h,H2氣氛400 ℃還原3 h,H2流速30 mL·min-1。所得催化劑為負載質量分數10%Ni/γ-Al2O3、10%Fe/γ-Al2O3和6%Ni-4%Fe/γ-Al2O3,分別標記為Ni/γ-Al2O3、Fe/γ-Al2O3和Ni-Fe/γ-Al2O3催化劑。
1.2 原位漫反射紅外光譜實驗
原位漫反射紅外光譜實驗采用HVC-DRP-3型高溫漫反射池,在德國布魯克公司TENSOR27型傅里葉變換紅外光譜儀上進行。將一定量的催化劑粉末裝入樣品池,并刮平樣品表面,以利于增強漫反射信號。
樣品池首先通入氦氣(流速20 mL·min-1)并逐漸升溫至350 ℃,恒溫30 min以吹盡催化劑表面的空氣。然后降至室溫,切換為常壓氫氣(流速為30 mL·min-1)并程序升溫(2 ℃·min-1)至400 ℃,在此溫度下還原催化劑1 h。還原結束后切換為氦氣吹掃30 min,降至室溫后切換為CO或體積分數0.5%CO-H2混合氣,控制流速為15 mL·min-1,使氣體在催化劑表面進行動態吸附,然后每隔一定時間進行紅外掃描,掃描次數為128次,分辨率為4 cm-1。
2 結果與討論
2.1 Fe/γ-Al2O3催化劑
圖1為不同溫度下Fe/γ-Al2O3催化劑上CO吸附的原位漫反射紅外光譜圖。由圖1可以看出,Fe/γ-Al2O3催化劑在2 174 cm-1和2 118 cm-1處出現氣相CO的吸收峰[19],除此之外,未觀察到其他紅外吸收峰。
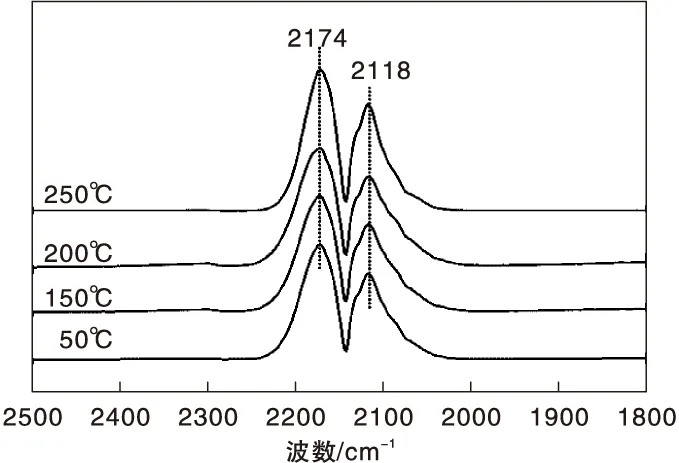
圖1 不同溫度下Fe/γ-Al2O3催化劑上CO吸附的原位漫反射紅外光譜圖Figure 1 In-situ DRIFTS of CO adsorbed on Fe/γ-Al2O3 catalyst at different temperatures
圖2為不同溫度下Fe/γ-Al2O3催化劑上CO-H2混合氣吸附的原位漫反射紅外光譜圖。與圖1相比,氣相CO的吸收峰強度明顯減弱,這是由于混合氣中CO的體積分數較小所致。此外,該催化劑上未觀察到其他任何吸收峰。顯然,無論H2存在與否,Fe/γ-Al2O3催化劑均未表現出CO吸附行為,這是其不具有CO甲烷化催化活性的直接原因。
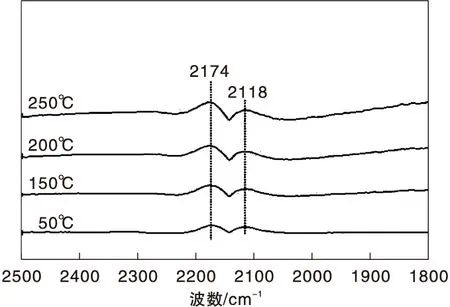
圖2 不同溫度下Fe/γ-Al2O3催化劑上CO-H2混合氣吸附的原位漫反射紅外光譜圖Figure 2 In-situ DRIFTS of CO and CO-H2 mixed gas adsorbed on Fe/γ-Al2O3 catalyst at different temperatures
2.2 Ni/γ-Al2O3催化劑
圖3為不同溫度下Ni/γ-Al2O3催化劑上CO吸附的原位漫反射紅外光譜圖。由圖3可以看出,50 ℃時Ni/γ-Al2O3催化劑表面除了氣相CO吸收峰外,在2 059 cm-1處還出現明顯的線式CO(COL)吸收峰[19-21]。隨著溫度升高,COL峰強度明顯增強,但在110 ℃后開始呈下降趨勢。200 ℃時,在2 359 cm-1和2 341 cm-1處出現氣相CO2的吸收峰,表明隨著溫度升高,部分化學吸附的CO發生歧化反應[22]。在(110~250) ℃,COL峰強度隨著溫度的升高逐漸減弱,表明CO在Ni/γ-Al2O3催化劑表面化學吸附變弱。
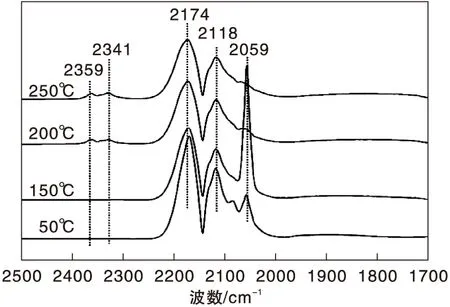
圖3 不同溫度下Ni/γ-Al2O3催化劑上CO吸附的原位漫反射紅外光譜圖Figure 3 In-situ DRIFTS of CO adsorbed on Ni/γ-Al2O3 catalyst at different temperatures
圖4為不同溫度下Ni/γ-Al2O3催化劑上CO-H2混合氣吸附的原位漫反射紅外光譜圖。
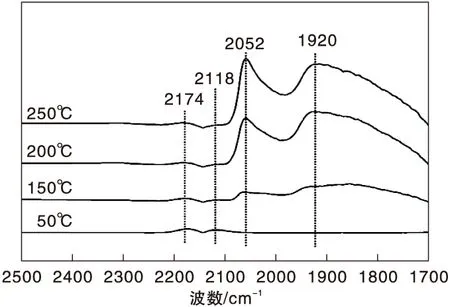
圖4 不同溫度下Ni/γ-Al2O3催化劑上CO-H2混合氣吸附的原位漫反射紅外光譜圖Figure 4 In-situ DRIFTS of CO-H2 mixed gas adsorbed on Ni/γ-Al2O3 catalyst at different temperatures
2.3 Ni-Fe/γ-Al2O3催化劑
圖5為不同溫度下Ni-Fe/γ-Al2O3催化劑上CO吸附的原位漫反射紅外光譜圖。由圖5可知,50 ℃時除了氣相CO吸附峰外,在2 057 cm-1處也出現微弱的COL吸收峰,且峰強度隨著溫度的升高逐漸增大,這與CO在Ni/γ-Al2O3催化劑上的吸附行為明顯不同,后者的COL吸收峰強度隨溫度升高先增大后減小。可見,與Ni/γ-Al2O3催化劑相比,Ni-Fe/γ-Al2O3催化劑表面存在較強的CO化學吸附中心,與Ni-Fe/γ-Al2O3催化劑中Ni-Fe合金的形成有關[18]。
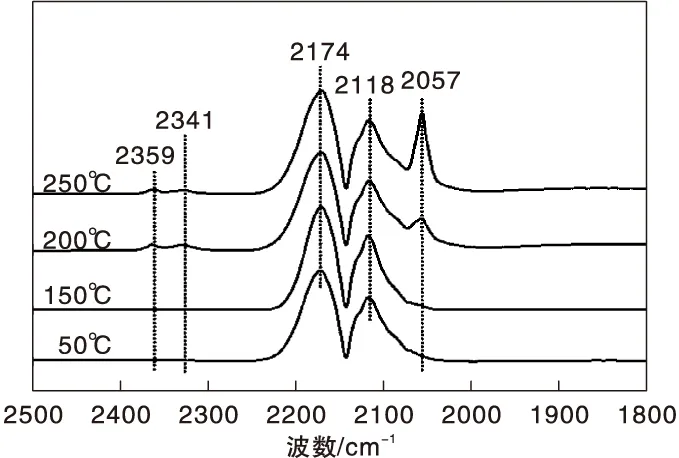
圖5 不同溫度下Ni-Fe/γ-Al2O3催化劑上CO吸附的原位漫反射紅外光譜圖Figure 5 In-situ DRIFTS of CO adsorbed on Ni-Fe/γ-Al2O3 catalyst at different temperatures
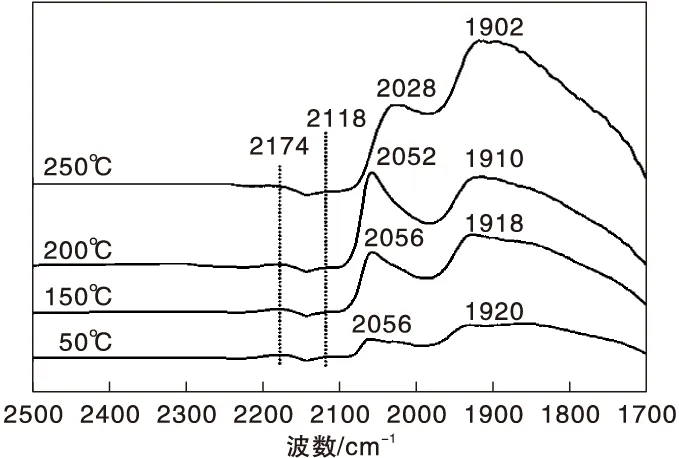
圖6 不同溫度下Ni-Fe/γ-Al2O3催化劑上CO-H2混合氣吸附的原位漫反射紅外光譜圖Figure 6 In-situ DRIFTS of CO-H2 mixed gas adsorbed on Ni-Fe/γ-Al2O3 catalyst at different temperatures
3 結 論
(1) 在CO吸附條件下,Fe/γ-Al2O3催化劑上未發生CO化學吸附,Ni/γ-Al2O3和Ni-Fe/γ-Al2O3催化劑上均有線式CO形成,但后者對CO的吸附能力明顯強于前者。
(2) 在CO-H2混合氣吸附條件下,Ni/γ-Al2O3催化劑表面以鎳羰基氫化物吸附為主,而Ni-Fe/γ-Al2O3催化劑表面則以鎳多氫羰基氫化物吸附為主,特別是橋式鎳多氫羰基氫化物。
(3) 鎳多氫羰基氫化物的Ni中心對CO分子反鍵π軌道更強的反饋電子能力導致Ni-C鍵增強,C-O鍵削弱,更有利于C-O鍵斷裂進而加氫進行甲烷化反應,因而Ni-Fe/γ-Al2O3催化劑顯示出更高的CO甲烷化催化活性。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
