大山楂顆粒的質量標準研究
2020-02-22 08:55:10杜成興劉明旭潘年瓊馮發進霍昕楊迺嘉
中國科技縱橫 2020年17期
關鍵詞:檢測
杜成興 劉明旭 潘年瓊 馮發進 霍昕 楊迺嘉
(1.貴州省生物技術研究開發基地,貴州貴陽 550002;2.貴州省科暉制藥廠,貴州清鎮 551400)
大山楂顆粒由山楂、六神曲、炒麥芽三味中藥組成,是根據2020年版《中國藥典》中的大山楂丸[1]處方改劑而成。大山楂顆粒的制劑方法與大山楂丸不同,大山楂丸是直接將藥材粉碎成細粉加入輔料制得的,大山楂顆粒是將藥材通過水煮后濃縮成浸膏再加入輔料制得的。丸劑釋藥緩慢,可延長藥效、降低毒性和刺激性,減少不良反應,適用于慢性病治療或病后調和氣血,但對于老人小孩及吞咽困難的人群來說不易服用;顆粒劑具有易溶解、易吸收、口感好的優點,更適合老人小孩及吞咽困難的人群服用[2-3]。處方中,山楂有降血壓、調血脂的作用,同時可保護肝臟,具有強心、抗氧化、提高免疫力的作用[4-5]。六神曲能夠消食化積、健脾和胃[6-8]。炒麥芽能行氣消食回乳,具有助消化、降血糖、抗真菌、抑制催乳素釋入等作用[9-10]。
在2020年版《中國藥典》中,大山楂丸是采用薄層色譜法檢測熊果酸的含量來測定的,但薄層色譜法存在一些缺陷,如受鋪板質量、點樣技術、展開條件、顯色效果等因素的影響,使測定結果誤差較大。高效液相色譜法比薄層色譜法更靈敏、簡單、快速、準確,且精密度好,能更好地控制藥物的質量。熊果酸是脂溶性成分,大山楂顆粒是山楂、六神曲、炒麥芽的水煎煮液濃縮成浸膏制備而成,經HPLC含量測定發現浸膏中熊果酸含量較低,而原兒茶酸的含量較高,且金絲桃苷、蘆丁、牡荊素等均不能鑒別出來,故選擇原兒茶酸作為指標成分進行檢測[11-14]。本研究采用高效液相色譜法對原兒茶酸進行含量測定,以期建立大山楂顆粒的質量標準[15],為大山楂顆粒的進一步開發利用提供參考。
1.儀器和材料
1.1 儀器
JJ20000B型電子天平(常熟市雙杰測試儀器廠),AUW120D型電子天平(島津儀器蘇州有限公司),AL204型電子天平(梅特勒—托利多儀器有限公司),ZF-90型多功能暗箱式紫外投射儀(上海山顧村電光儀器廠),Ultimate 3000高效液相色譜儀(美國賽默飛公司),SPD-16C 高效液相色譜儀(島津儀器蘇州有限公司),GZX-9140MBE電熱鼓風干燥箱(上海博訊實業有限公司醫療設備廠)。其他儀器均由貴州省生物技術研究開發基地提供。
1.2 材料
熊果酸對照品(批號:110742-200314)、原兒茶酸對照品(批號:110809-201205)、金絲桃苷對照品(批號:111521-201809)、蘆丁對照品(批號:100080-201610)、牡荊素對照品(批號:11687-201704)、山楂對照藥材(批號:121138-201606)均采購于中國食品藥品檢定研究院。大山楂顆粒樣品、藥材及試劑由貴州省生物技術研究開發基地提供。
2.方法和結果
2.1 大山楂顆粒的TLC定性鑒別
取大山楂顆粒研細,稱取3g,加入50%乙醇2ml潤濕,再加入乙酸乙酯10ml,超聲30min,過濾,濾液揮干,加入乙酸乙酯1ml溶解,得供試品溶液;取山楂對照藥材3g,同法制得山楂對照藥材溶液;取缺山楂陰性樣品研細,稱取3g,同法制得缺山楂陰性樣品溶液。吸取上述三種溶液各5μL,分別點于同一聚酰胺薄膜板上,以乙酸乙酯—環己烷—甲酸(10:3:1)為展開劑,展開,取出晾干,噴以0.1%溴甲酚綠(溴甲酚綠0.1g,溶于100ml乙醇中,加1%NaOH溶液至藍色即可),吹至斑點清晰,結果見圖1。TLC圖譜顯示,大山楂顆粒在與山楂對照藥材相應位置上顯相同顏色的斑點,且顯色斑點清晰,陰性無干擾。
取15批大山楂顆粒研細,各稱取3g,同法測定,結果見圖2。結果表明,十五批大山楂顆粒供試品在與山楂對照藥材相應的位置上,顯相同的黃色斑點,且斑點清晰,陰性無干擾。

圖1 山楂TLC色譜圖
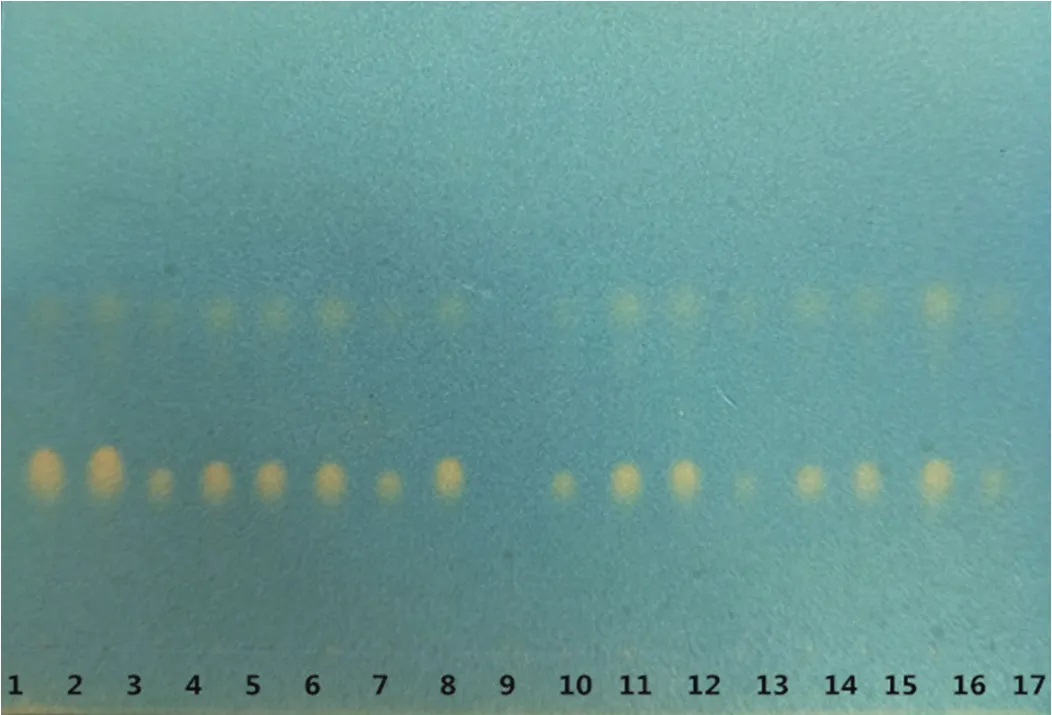
圖2 十五批大山楂顆粒的TLC色譜圖
2.2 高效液相色譜法
2.2.1 色譜適應性條件
依利特C18色譜柱(250mm×4.6mm,5μm);流動相為乙腈-1%醋酸水(5:95);流速0.9ml/min;檢測波長260nm;柱溫30℃;進樣量10μL。結果表明,在此色譜條件下,原兒茶酸峰形較好,約9min左右出峰,理論塔板數高于3000。
2.2.2 樣品制備方法
取大山楂顆粒磨成細粉,稱取樣品1.0g,置于具塞錐形瓶中,加入0.1mol/L鹽酸2ml潤濕,再加入20ml甲醇-1%醋酸水(7:3),超聲提取30min,過濾,加甲醇-1%醋酸水(7:3)定容到25ml,得樣品溶液。
2.2.3 對照品制備方法
精密稱取原兒茶酸對照品9.82mg,置于10ml容量瓶中,加入甲醇-1%醋酸水(7:3)溶液定容至刻度線;再從中精密量取1ml于50ml容量瓶中,加入甲醇-1%醋酸水(7:3)溶液定容至刻度線,得到原兒茶酸對照品溶液(19.64μg/ml)。
2.2.4 指標性成分篩選實驗
按上述色譜條件對大山楂顆粒中原兒茶酸進行檢測,結果見圖3。結果表明在大山楂顆粒中檢出原兒茶酸,但在炒麥芽藥材中沒有檢測出,故選擇原兒茶酸作為指標性成分。
2.2.5 方法學考察
(1)線性關系。精密配制6種濃度的原兒茶酸對照品溶液:2.455μg/ml、4.91μg/ml、9.82μg/ml、19.64μg/ml、39.28μg/ml、78.56μg/ml,分別進樣 10μL,以原兒茶酸濃度為X軸,峰面積為Y軸,得線性回歸方程。結果表明,線性回歸方程為y=42756x+16583(r=0.9999),原兒茶酸在2.455μg/mL~78.56μg/mL范圍內呈現良好的線性關系。
(2)精密度實驗。按2.2.1項下色譜條件,取同一濃度的原兒茶酸對照品溶液(19.64μg/ml),進樣6次,每次10μL,以原兒茶酸對照品溶液(19.64μg/ml)為參照,計算樣品中原兒茶酸的含量。結果顯示,RSD=0.78%,符合中國藥典要求(RSD<2%),表明HPLC儀器精密性良好。
(3)中間精密度實。按2.2.1項下色譜條件,取同一濃度的原兒茶酸對照品溶液(19.64μg/ml),進樣6次,每次10μL,以原兒茶酸對照品溶液(19.64μg/ml)為參照,計算樣品中原兒茶酸的含量。結果顯示RSD=1.06%,符合《中國藥典》要求(RSD<2%),表明中間精密度良好。
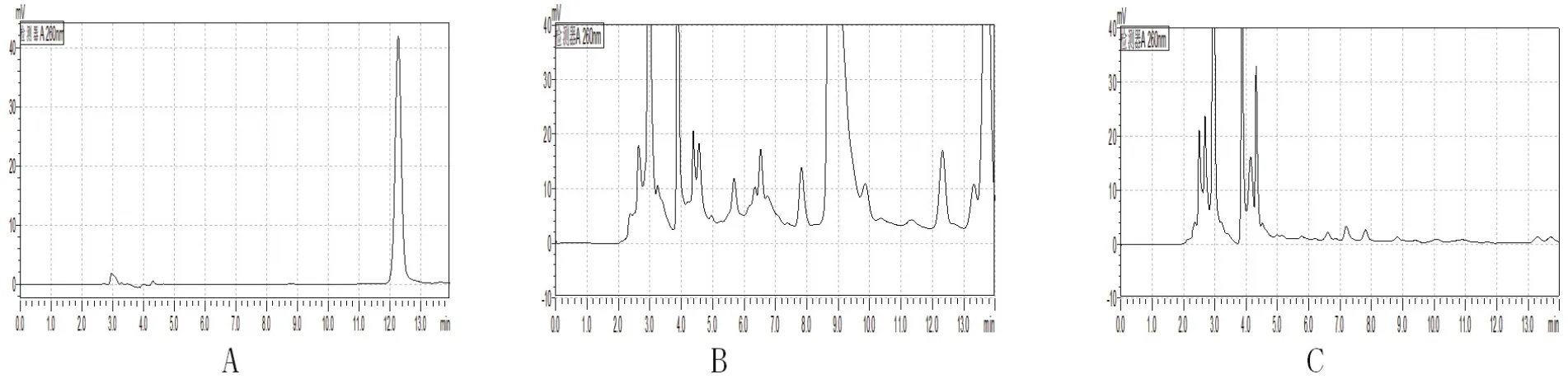
圖3 原兒茶酸HPLC色譜圖
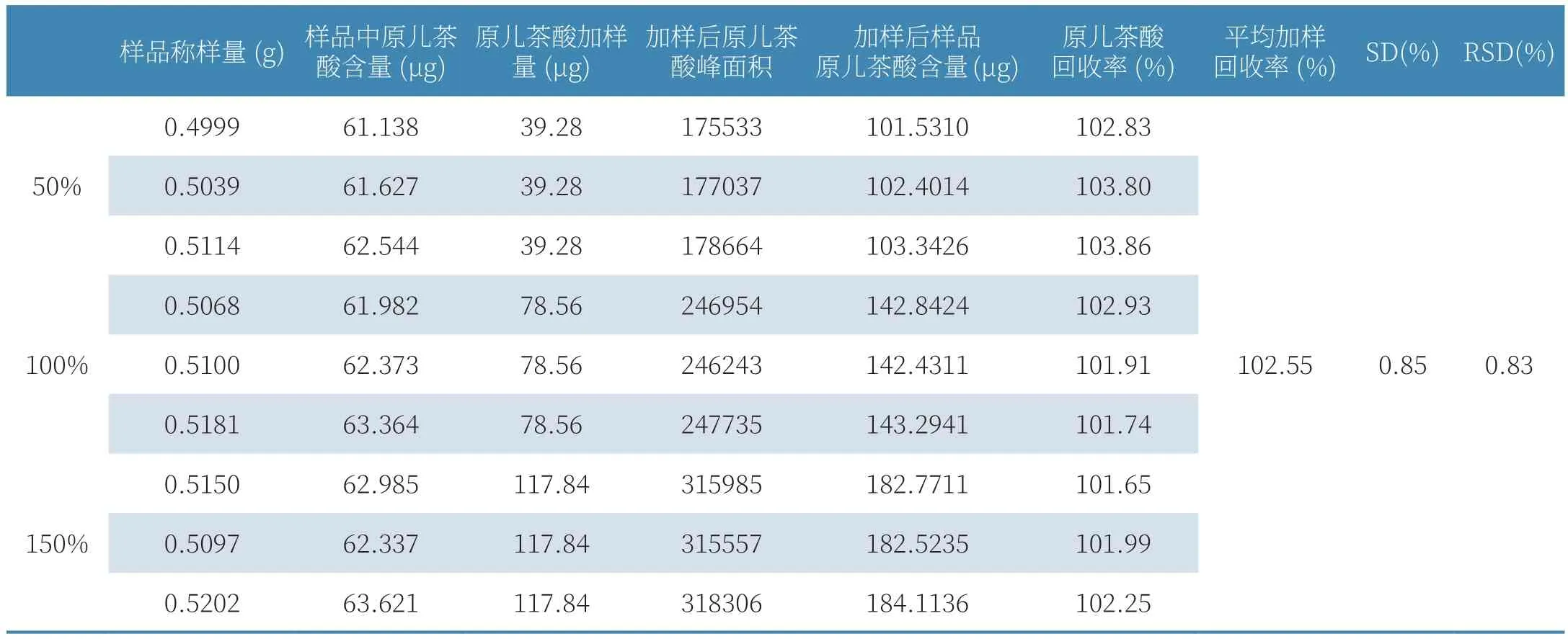
表1 加樣回收率實驗結果
(4)重復性實驗。取大山楂顆粒磨成細粉,精密稱取6份,每份1.0g。按2.2.2項下制備方法,得到樣品溶液,按2.2.1項下色譜條件,進樣6次,每次10μL,以原兒茶酸對照品溶液(19.64μg/ml)為參照,計算樣品中原兒茶酸的含量。結果顯示RSD=0.33%,符合《中國藥典》要求(RSD<2%),表明實驗重復性良好。
(5)穩定性實驗。取大山楂顆粒磨成細粉,精密稱取1.0g。按2.2.2項下制備方法,得樣品溶液,間隔1h進樣一次,按2.2.1項下色譜條件,進樣6次,每次10μL,以原兒茶酸對照品溶液(19.64μg/ml)為參照,計算樣品中原兒茶酸的含量。結果顯示RSD=0.66%,符合中國藥典要求(RSD<2%),表明供試品在6h內穩定。
(6)加樣回收率實驗。取大山楂顆粒磨成細粉,精密稱取9份,每份1.0g,分為3組。每組按取樣量的50%、100%、150%分別加入原兒茶酸對照品,按2.2.2項下制備方法,得樣品溶液,按2.2.1項下色譜條件,進樣9次,每次10μL,以原兒茶酸對照品溶液(19.64μg/ml)為參照,計算樣品中原兒茶酸的含量,結果見表1。結果顯示,平均加樣回收率結果為102.55%,RSD=0.83%,符合《中國藥典》要求(RSD<2%),表明實驗可行性良好,該方法可用于大山楂顆粒的含量測定。
2.2.6 含量測定
取15個不同批次的大山楂顆粒磨成細粉,每份稱取1.0g,按2.2.2項下制備方法,得樣品溶液,按2.2.1項下色譜條件,進樣6次,每次10μL,以原兒茶酸對照品溶液(19.64μg/ml)為參照,計算樣品中原兒茶酸的含量,結果見表2。結果表明:大山楂顆粒中原兒茶酸的平均含量為0.0653mg/g,最低為0.0265mg/g。
3.結論與討論
由于大山楂顆粒是通過水煎煮而得的,且炒麥芽與六神曲中存在許多酶類,所以在煎煮過程中,許多的成分都通過酶反映了在煎煮過程中有許多成分可能會發生變化。通過多次的試驗發現,在處方藥材中能夠檢測出來的許多成分在樣品中不能被檢測出來,而樣品中能被檢測出的成分在藥材中有不能被檢測出來。在高效液相色譜法測定山楂含量中,對已有山楂報道中的很多成分檢測后發現,在樣品中都不能被檢測出來,經過很多次試驗發現原兒茶酸在藥材和樣品中都被檢測出來,通過試驗得出藥材與樣品的轉化率均大于100%(三批樣品的轉化率分別是113%、137%、146%),說明原兒茶酸在煎煮的過程中由于酶的作用生成了更多的原兒茶酸。因此,最低限量不是通過轉移率來計算,而是通過15批最低量再低一點來確定。
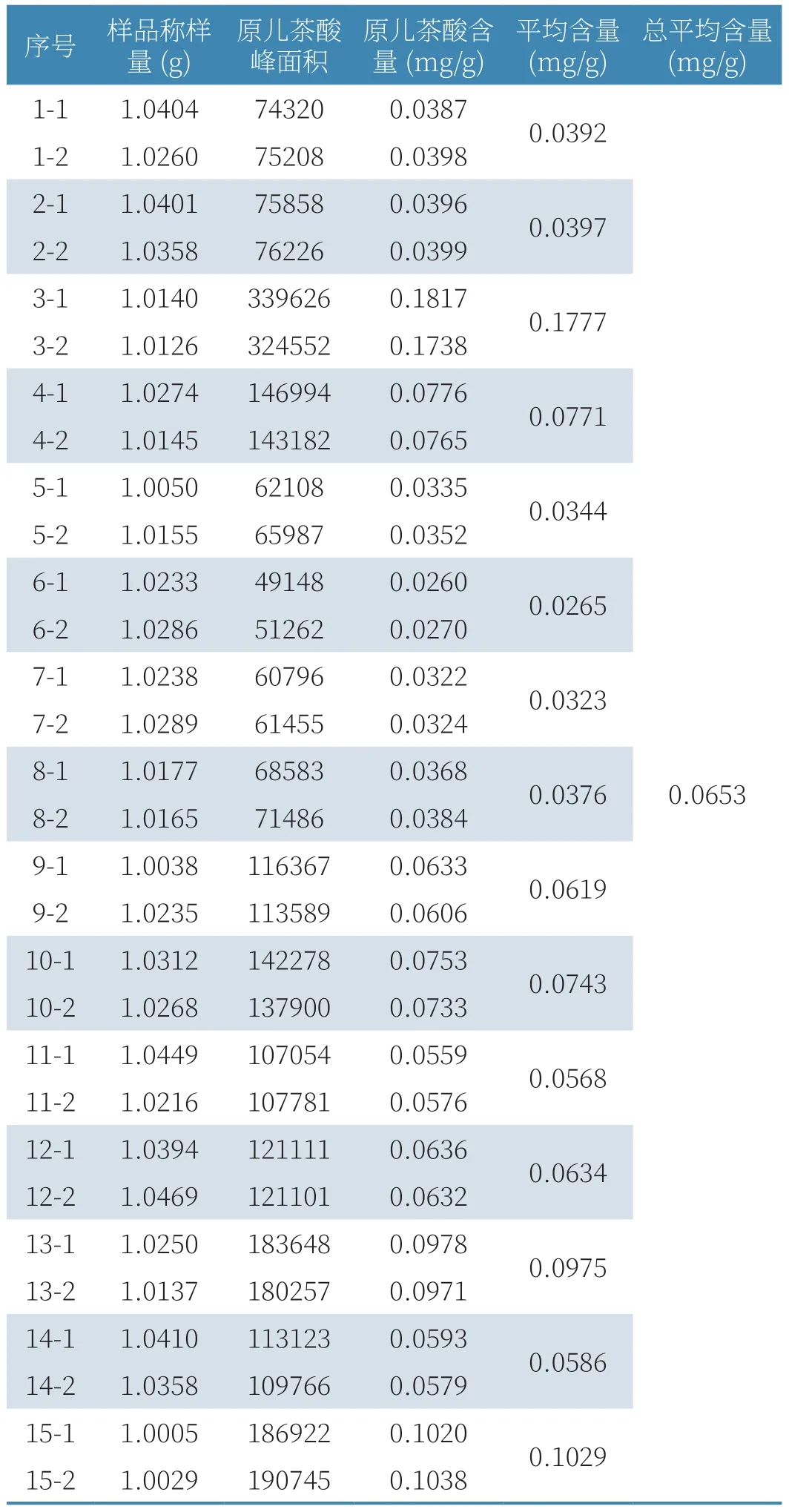
表2 含量測定實驗結果
對15批大山楂顆粒樣品的含量測定發現,大山楂顆粒的平均含量為0.0653mg/g,最低含量為0.0265mg/g,考慮到藥材每年含量會有所不同,因此將大山楂顆粒中的原兒茶酸的限量定為:大山楂顆粒每包含原兒茶酸不得少于50μg。另外,大山楂顆粒的薄層色譜鑒別中還有六神曲和炒麥芽都沒有被鑒別出來。含量測定方面,本研究沒能找出單一的一味藥的含量測定,只是控制了大山楂顆粒中原兒茶酸的總含量。穩定性研究方面,也只是做了初步的研究,對于大山楂顆粒還需要做更長期的穩定性研究。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
