利用重組大腸桿菌發酵生產L-茶氨酸
2020-01-14 02:33:44張通龍科藝曹華杰吳思佳元躍陳寧范曉光
食品與發酵工業 2019年22期
張通,龍科藝,曹華杰,吳思佳,元躍,陳寧,3,范曉光,3*
1(天津科技大學 生物工程學院,天津,300457)2(河南巨龍生物工程股份有限公司,河南 汝州,467500) 3(天津市微生物代謝與發酵過程控制技術工程中心,天津,300457)
L-茶氨酸是茶葉中特有的氨基酸,其質量分數占其總游離氨基酸50%以上[1]。L-茶氨酸有許多有益的生理效果,能夠促進人腦放松,提高注意力和促進學習能力[2]。 在醫療保健方面,L-茶氨酸具有降低血壓、預防血管疾病、緩解壓力、保護神經、減肥降脂和提高免疫系統能力等功效[3-9]。在食品方面,L-茶氨酸已被FDA列為安全的食品藥品添加劑,用于改良食品品質、增強食品風味[10]。
現有的L-茶氨酸的生產方法主要包括提取法、化學合成法和酶催化法。提取法是最天然的L-茶氨酸生產方法,但由于干茶葉中L-茶氨酸的質量分數僅占1%~2%,導致從茶葉或茶葉廢渣中提取L-茶氨酸提取量少、產品純度低,不能滿足大規模生產需求[11]。化學合成法是目前茶氨酸生產的主要途徑,是利用酯型或乙酰化的L-谷氨酸和乙胺作為底物,通過控制反應過程和反應條件使L-谷氨酸和乙胺結合形成L-茶氨酸[12],但由于合成產物易存在對映異構體,導致分離困難、產品收率低。近來使用酶催化法生產L-茶氨酸引起了很多學者的關注,目前主要有4種不同的細菌來源的酶可作為理想的生物催化劑并顯示出不同的潛力。這4種細菌來源的酶,包括L-谷氨酰胺合成酶(L-glutamine synthetase, GS)[13],γ-谷氨酰甲基酰胺合成酶(γ-glutamylmethylamide, GMAS)[14],γ-谷氨酰轉肽酶(γ-glutamyltranspeptidase, GGT)[15]和L-谷氨酰胺酶[16]。上述4種細菌酶都被證實了有較高的催化L-茶氨酸合成的活性,但這些已被證實的酶的微生物來源十分有限,且酶催化法合成L-茶氨酸包括產酶菌株的培養和酶促反應兩個過程,需要使用價格較高的谷氨酸/谷氨酰胺鹽和ATP為原料[17],生產成本與提取法和化學法相比并無優勢。
鑒于現有L-茶氨酸生產方法的不足,本研究通過構建重組大腸桿菌高效表達來源于Methylovorusmays的γ-谷氨酰甲基酰胺合成酶,并通過流加前體物乙胺發酵的方式直接發酵合成L-茶氨酸。根據L-茶氨酸的物化性質擬定出合適的分離提取路線,從發酵液中獲得了合格的L-茶氨酸成品。整個生產工藝具有較好的經濟可行性和工業應用潛力。
1 材料和方法
1.1 材料與試劑
1.1.1 菌株和質粒
實驗所需菌株和質粒如表1所示。

表1 菌株和質粒Table 1 Strains and plasmids
1.1.2 培養基
斜面培養基(g/L):葡萄糖 1,蛋白胨 10,牛肉膏 10,酵母粉 5,NaCl 2.5,瓊脂 25,pH 7.0;
種子培養基(g/L):葡萄糖 25,酵母提取物 10,蛋白胨 15,NaCl 15,pH 7.2;
發酵培養基(g/L):葡萄糖30,酵母提取物10,蛋白胨15,KH2PO45,MgSO42,pH 7.2。
1.1.3 主要試劑
PCR產物回收及質粒提取試劑盒:北京博大泰克生物基因有限責任公司;L-茶氨酸標品:美國Sigma公司;其他試劑均為國產分析純。
1.2 目的基因擴增
1.2.1 以DNA為模板的PCR
在PCR儀上根據引物和片段長度分別設定退火溫度(約低于理論退火溫度5 ℃)和延伸時間。本實驗使用Prime STAR酶作為DNA聚合酶,擴增速率約為1 kb/min。PCR反應條件:首先95 ℃預變性5 min,之后進入如下循環:95 ℃變性30 s,55 ℃退火30 s,72 ℃延伸合適時間(根據片段長度),完成最后一個循環后,72 ℃繼續延伸10 min。其中驗證體系進行25個循環,回收體系進行30個循環。擴增PCR反應液體系如表2所示。

表2 擴增PCR反應液體系Table 2 Composition of PCR reaction solution
1.2.2 重疊PCR
以T7-gmas基因整合為例。根據GenBank中E.coliW3110的yghX基因序列、T7啟動子和Methylovorusmays的gmas序列,在Primer Premier 5.0中設計引物,分別擴增上游同源臂、下游同源臂和中間整合片段。其中相連片段分別有約20 bp的重疊區域。PCR擴增體系如表3所示。

表3 重疊PCR擴增體系Table 3 Amplification system of overlap PCR
分別回收T7-gmas上、下游同源臂和整合片段PCR產物,以1∶1∶2的摩爾比進行混合作為模板,通過重疊PCR,將上游、下游和中間整合片段3段片段連接起來。
1.3 CRISPR/Cas9基因整合方法
1.3.1 帶有擬敲除基因同源臂基因片段的擴增
帶有擬敲除基因同源臂片段擴增的方法參見1.2.1以DNA為模板的PCR和1.2.2重疊PCR。PCR擴增完成后切膠回收片段。
1.3.2 整合基因替換擬敲除目的基因
將重疊片段和gRNA質粒共同電轉入含有pRedCas9質粒的大腸桿菌細胞感受態中,于32 ℃培養,待長出單菌落,經菌落PCR鑒定篩選出陽性轉化子,再消除pGRB和pRedCas9。
1.4L-茶氨酸的發酵生產方法
從-80 ℃冰箱保菌管中刮1環菌種,均勻涂布于活化斜面,37 ℃培養12 h,轉接茄形瓶繼續培養12 h。取適量無菌水于茄形瓶中,將菌懸液接入種子培養基中,pH值穩定在7.0左右,溫度恒定在37 ℃,溶氧在20%~30%,培養6 h。按照15%接種量接入新鮮的發酵培養基,開始發酵,發酵過程中控制pH值穩定在7.0左右,溫度維持在37 ℃,溶氧在25%~35%之當培養基中的葡萄糖消耗完之后,流加800 g/L的葡萄糖溶液并維持發酵培養基中的葡萄糖濃度<2 g/L,發酵開始4 h時開始以30 mL/h的速率流加2 mol/L的乙胺溶液至發酵結束。
1.5L-茶氨酸的分離純化
根據L-茶氨酸的物化性質制定了合理的產品分離提取工藝。具體流程為:微濾去除發酵液中的菌體,微濾膜的孔徑為0.1 μm,料液循環體積為0.2 L,過濾流速為0.5~10 L/h;超濾膜除蛋白質和色素,超濾膜截留分子質量600 Da;初步濃縮脫色,使用陽離子交換樹脂進行濃縮脫色;藥用炭脫色,用量3%(質量分數)的活性炭,溫度60 ℃,攪拌混合時長25 min;二次濃縮,使用真空濃縮裝置,真空度-0.1 MPa,濃縮至飽和;冷乙醇析晶;重結晶,將結晶用去離子水溶解,然后重復濃縮至冷乙醇析晶的步驟;利用減壓烘干設備,烘干成品,成品含水質量分數<5%。測定膜過濾處理前后溶液在600 nm下的吸收值,根據公式(1)即可求得菌體細胞截留率Ycell;測定膜過濾和脫色處理前后溶液在540 nm下的吸收值,根據公式(2)即可求得色素截留率Ypigment;根據考馬斯亮藍顯色方法測定可溶性蛋白質含量,根據公式(3)可以求得蛋白截留率Yprotein:
(1)
(2)
(3)
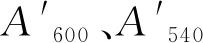
1.6L-茶氨酸的檢測方法
發酵液預處理:取1 mL發酵液于1.5 mL EP管中,13 000 r/min離心2 min,取上清液待測。
衍生處理:用移液器取200 μL衍生緩沖液加入到2 mL離心管中,取上述上清液10 μL加入反沖液中,再加入300 μL衍生劑溶液,充分混勻后,將反應液置于65 ℃水浴鍋中避光反應1 h,取出加入定容緩沖液定容至1.2 mL,充分搖勻后過0.2 μm膜后進行色譜分析[18]。
高效液相色譜檢測:色譜柱為Agilent ZORBAX Eclipse AAA(4.6 mm×150 mm,5-Micron),流動相為乙酸鈉緩沖液,50%乙腈,柱溫33 ℃,流速1 mL/min,檢測波長360 nm,采用二元梯度分析。
2 結果與分析
2.1 在大腸桿菌E. coli origin中單拷貝T7啟動子控制的γ-谷氨酰甲胺合成酶基因gmas
以E.coliorigin基因組為模板,根據其yghX基因的上下游序列設計上游同源臂引物UP-yghX-S、UP-yghX-A和下游同源臂引物DN-yghX-S、DN-yghX-A,通過PCR擴增其上下游同源臂片段。根據針對大腸桿菌優化合成的gmas基因序列,設計引物gmas-S、gmas-A,通過PCR擴增gmas基因片段,將啟動子T7設計在上游同源臂的下游引物和gmas基因的上游引物中。通過重疊PCR將上述片段整合為“上游同源臂-T7-gmas-下游同源臂”的重疊片段。設計引物gRNA-yghX-S和gRNA-yghX-A擴增包含靶序列的DNA片段,與線性化的pGRB載體重組后獲得重組的pGRB-yghX。將整合片段和pGRB-yghX電轉化至含有pREDCas9載體的E.coliorigin感受態細胞中,將電轉化后復蘇培養的菌體涂布于含氨芐青霉素和奇霉素的LB平板上,32 ℃過夜培養后利用PCR驗證陽性重組子,再消除用于基因編輯的pGRB-yghX。
圖1是T7-gmas整合片段的陽性菌株PCR驗證的電泳圖。其中,上游同源臂的長度應為648 bp,gmas基因片段長度應為1 415 bp,下游同源臂的長度應為595 bp,整合片段的總長應為2 607 bp,PCR驗證時,陽性菌PCR擴增片段長度應為2 607 bp,原菌PCR擴增片段長度應為1 765 bp。電泳圖證明該片段整合成功。將單拷貝T7-gmas基因的菌株命名為E.coligmas1。

圖1 yghX::T7-gmas的PCR驗證圖Fig.1 PCR verification diagram of yghX::T7-gmas注:M:1kb Marker;1:上游同源臂;2:目的基因;3:下游同源臂;4:重疊片段;5:原菌PCR片段;6:目的菌PCR片段。圖2同。
2.2 在大腸桿菌E. coli origin中雙拷貝T7啟動子控制的γ-谷氨酰甲胺合成酶基因gmas
以E.coliorigin基因組為模板,根據其yeeP基因的上下游序列設計上游同源臂引物UP-yeeP-S、UP-yeeP-A和下游同源臂引物DN-yeeP-S、DN-yeeP-A,通過PCR擴增其上下游同源臂片段。根據針對大腸桿菌優化合成的gmas基因序列,設計引物gmas-S、gmas-A,通過PCR擴增gmas基因片段,將啟動子T7設計在上游同源臂的下游引物和gmas基因的上游引物中。通過重疊PCR將上述片段整合為“上游同源臂-T7-gmas-下游同源臂”的重疊片段。設計引物gRNA-yeeP-S和gRNA-yeeP-A擴增包含靶序列的DNA片段,與線性化的pGRB載體重組后獲得重組pGRB-yeeP。將整合片段和pGRB-yeeP電轉化至含有pREDCas9載體的E.coliorigin感受態細胞中,將電轉化后復蘇培養的菌體涂布于含氨芐青霉素和奇霉素的LB平板上,32 ℃過夜培養后利用PCR驗證陽性重組子,再消除用于基因編輯的pGRB-yeeP。
圖2是T7-gmas整合片段的陽性菌株PCR驗證的電泳圖。其中,上游同源臂的長度應為558 bp,gmas基因片段長度應為1 415 bp,下游同源臂的長度應為547 bp,整合片段的總長度應為2 469 bp。PCR驗證時,陽性菌PCR擴增片段長度應為2 469 bp,原菌PCR擴增片段長度應為1 396 bp。電泳圖證明該片段整合成功。將雙拷貝T7-gmas基因的菌株命名為E.coligmas2。
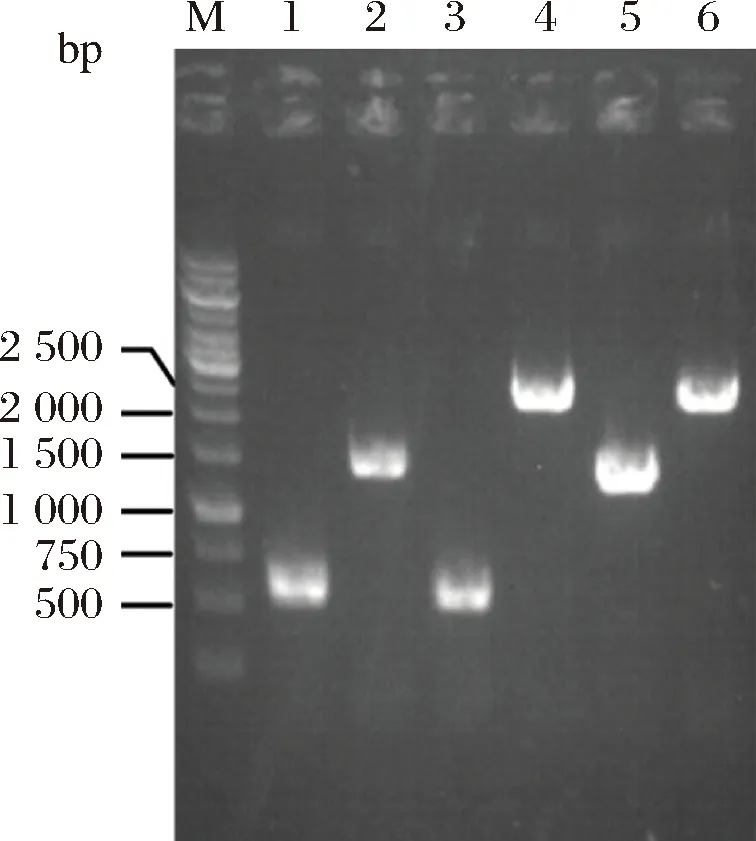
圖2 yeeP::T7-gmas的PCR驗證圖Fig.2 PCR verification diagram of yeeP::T7-gmas
2.3E. coli gmas1和E. coli gmas2的5 L發酵罐發酵結果比較
為對比2株菌生產L-茶氨酸的能力,對單拷貝E.coligmas1和雙拷貝E.coligmas2菌株進行了5 L發酵罐發酵實驗,由于大腸桿菌能夠通過自身代謝產生L-谷氨酸和ATP,因此只需要在發酵過程中外源添加前體物乙胺即可獲得L-茶氨酸。結果如圖3所示。由圖3可知,發酵結束時,E.coligmas2菌株5 L發酵罐發酵L-茶氨酸產量為30.45 g/L,比E.coligmas1提高了22%左右,而菌體濃度E.coligmas2菌株比E.coligmas1降低了11%左右。在耗糖量方面,E.coligmas2菌株的耗糖量為150.97 g/L,而E.coligmas1菌株為124.17 g/L,兩者的糖酸轉化率均為20%左右。上述結果表明增加γ-谷氨酰甲胺合成酶基因gmas的拷貝數雖然能夠提升L-茶氨酸的產量,但對糖酸轉化率影響不大,且過多的異源基因表達會加重菌體的生長負擔。后期可考慮通過進一步強化葡萄糖到L-谷氨酸的代謝流量來提升糖酸轉化率,從而增加葡萄糖的利用率。
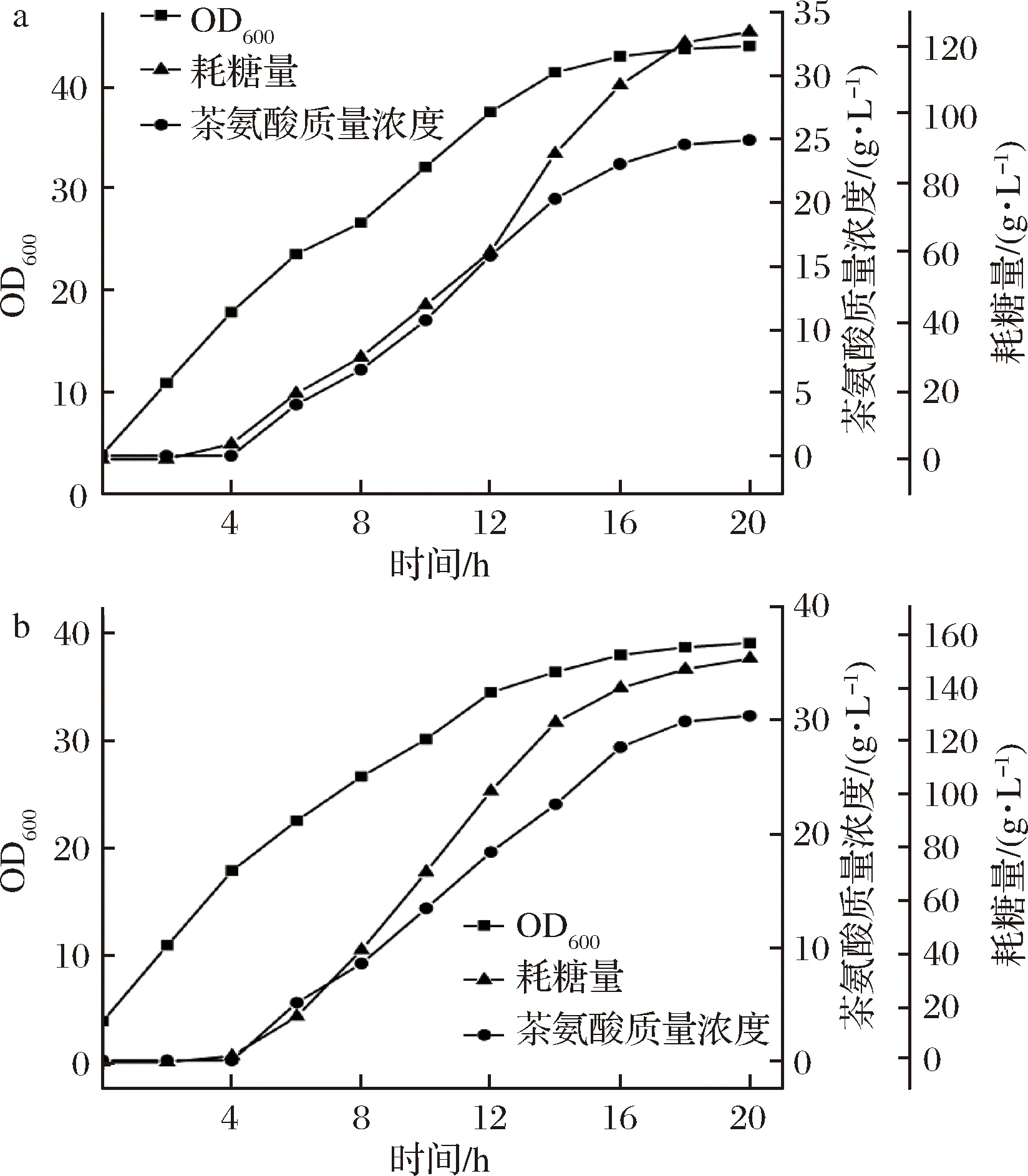
a-E. coli gmas1;b-E. coli gmas2圖3 5 L發酵罐中不同重組菌發酵L-茶氨酸的進程曲線Fig.3 Fermentation process curve of L-theanine in 5 L-fermetor by different recombinant strain
2.4 從發酵液中分離提取L-茶氨酸產品
發酵完成后,為了得到L-茶氨酸產品需要對發酵液進行分離提取。與大多數發酵液一樣,L-茶氨酸的發酵液中殘留有菌體、雜蛋白以及未被利用的培養基等雜質。因此合適的分離提取工藝就顯得尤為重要。本實驗首先根據L-茶氨酸的理化性質和發酵液的性質制定了如圖4所示的分離提取工藝流程。該流程主要將膜過濾,陽離子交換樹脂脫色濃縮,活性炭脫色和濃縮結晶四部分單元操作結合起來分離提取L-茶氨酸。
圖4 從發酵液中分離提取L-茶氨酸的流程圖Fig.4 Flow chart of the L-theanine separation and purification
發酵液成分復雜,黏度較大,固液共存,首先要經過初步處理去除其中的菌體以及可溶性蛋白和色素等大分子物質。常用的去除菌體的方法包括離心法,絮凝法和膜過濾法[19]。微濾膜能有效分離0.1~10 μm的粒子,操作簡便,過濾效率高,不會造成產品污染,適合用于L-茶氨酸發酵液的菌體分離;超濾法操作簡便,安全性能高,常用于分離分子質量>5 kDa物質,因此更適合用于該單元操作。據此本實驗選擇微濾膜和超濾膜組合的方式對發酵液中L-茶氨酸進行初步分離提取。
L-茶氨酸發酵液經過膜過濾后有些色素還是無法去除,這將會影響下一步的分離純化,并導致產品質量變差,所以需要進行進一步脫色處理才能進行后面的操作。目前,常用的脫色方法主要有離子交換樹脂脫色和活性炭脫色。離子交換法具有效率高、操作簡單、可重復利用等優點,是工業上常用的濃縮和脫色方法之一[20];活性炭脫色具有操作工藝簡單、設備投入少以及運行費用低等優點[21],其中藥用活性炭具有脫色速度快、吸附能力強、內部孔隙發達、孔隙粗大等特點,能夠有效吸附產品溶液中的色素,同時降低其中雜質,不影響產品其他成分濃度[22]。由于L-茶氨酸結構與L-谷氨酸較為接近,因此在乙醇中的溶解度較小,再結晶過程中使用冷乙醇可以增加晶體的析出率。重結晶操作是為了進一步提高產品的純度,去除析晶過程中包裹的雜質。
為了對比分離提取工藝中各步驟的分離效果,利用1.5中的公式對各步工藝中的細胞截留率Ycell、色素截留率Ypigment、蛋白截留率Yprotein和L-茶氨酸回收率進行了計算,各步工藝分離效果如表4所示。微濾和超濾膜聯用能夠截留99.5%的細胞和95.5%的可溶性蛋白,但對于色素的截留效果只有71.7%。陽離子交換樹脂結合藥用炭處理能夠截留98.7%的色素,使得產品能夠以白色晶體形式析出。整個過程L-茶氨酸提取回收率可達到70.34%,經高效液相色譜檢測成品純度可達到98.51%。
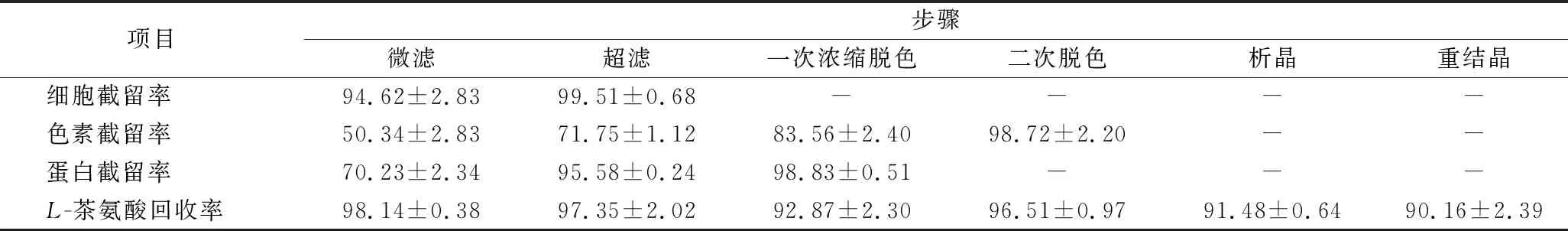
表4 分離提取工藝各步驟分離效果對比 單位:%
注:“-”表示無。
使用核磁共振對烘干后成品進行進一步鑒定,圖5為產品的核磁共振氫譜圖,其中4.713 ppm處為試劑峰。由圖中峰面積和峰數目可知,該產品共含有1個甲基、1個次甲基和3個不同的亞甲基。由峰位移和峰裂分數可知,上述每類基團所處位置,將圖上的峰從右至左依次排列順序,得到位置如圖5所示。由偶合常數J可知該產品構型為L型。之后與樣品核磁共振氫譜圖比對,確認產品為L-茶氨酸。

圖5 L-茶氨酸產品的氫譜核磁共振圖Fig.5 NMR hydrogen spectrum of L-theanine product
3 結論
L-茶氨酸作為一種重要的食品添加劑和藥品添加劑,已經廣泛應用于食品和醫藥領域。然而,現有的L-茶氨酸生產方法如提取法、化學合成法和酶催化法存在提取收率低、工藝復雜、生產成本高等不足。本研究通過Crispr/Cas9技術在大腸桿菌染色體上雙拷貝來自于Methylovorusmays的γ-谷氨酰甲基酰胺合成酶基因gmas,得到的重組菌E.coligmas2經過20 h乙胺流加發酵,L-茶氨酸產量可以達到30.45 g/L,糖酸轉化率可以達到20.17%,與現有的生產方法相比具有原料價格低廉、周期短、操作簡便、綠色環保等優點,工業應用價值較高。根據L-茶氨酸的物化性質擬定出合適的分離提取路線,獲得了合格的L-茶氨酸產品,整個過程提取回收率可達到70.34%,成品純度可達到98.51%。本研究證實了使用重組大腸桿菌直接發酵法生產L-茶氨酸是可行的,但是L-茶氨酸發酵水平還有待提高。后期可以針對前體物L-谷氨酸合成涉及的糖酵解途徑以及三羧酸循環中的關鍵代謝節點進行系統改造,進一步增強α-酮戊二酸至L-谷氨酸的代謝流,進而提高L-茶氨酸的產量及轉化率。
