先天性腎上腺皮質增生癥17α-羥化酶缺陷癥1例報告
2019-12-20 00:58:40高藝洋劉苗李靜單忠艷
中國醫科大學學報 2019年12期
高藝洋,劉苗,李靜,單忠艷
(中國醫科大學附屬第一醫院內分泌與代謝病科,內分泌研究所,沈陽 110001)
先天性腎上腺皮質增生癥(congenital adrenal hyperplasia,CAH)是由基因缺陷所致腎上腺皮質類固醇激素合成酶中某種或數種酶先天性活性缺乏引起的一組常染色體隱性遺傳性疾病。CAH種類繁多,其中17α-羥化酶缺陷癥(17α-hydroxylase deficiency,17α-OHD)類型相對少見,約占1%。2012年我院收治了1例17α-OHD患者,現將其診治情況報道如下:
1 臨床資料
患者,女,16歲,遼寧省人。2012年11月21日以“月經至今未來潮”為主訴入院就診。患者自訴至青春期時仍無月經來潮,父母非近親婚配。測血壓最高150/95 mmHg,伴有低鉀血癥。患者平素常自覺乏力、手腳麻木,休息數分鐘后可自行緩解。5~6歲時曾間斷出現手指、腳趾、口周及舌尖麻木,未在意。10歲時曾患腦炎,伴有右側肢體活動不靈及血鉀較低,于中國醫科大學附屬盛京醫院治療好轉后出院。
1.1 體格檢查
患者身高157 cm,體質量43 kg,指間距155 cm,上部量72 cm,下部量85 cm。無胡須生長,無毳毛增多,前額發際無退縮,無滿月臉及多血質面容,齒齦有輕度色素沉著,未見喉結,頸部無假性黑棘皮,無腋毛生長,無頸蹼及肘外翻。雙乳腺Tanner1期(圖1),無溢乳。掌紋未見色素沉著,下腹部及大腿內外側未見紫紋,雙下肢無浮腫,陰毛Tanner1期(圖2),外陰呈女性分布,無陰蒂肥大。手指、腳趾無畸形。四肢肌力Ⅴ級。雙上肢血壓均為140/90 mmHg,心率85次/min,未聞及病理性雜音。
圖1 患者乳房發育不良

圖2 患者外陰發育呈女性幼稚型
1.2 實驗室及影像學檢查
血 鉀3.28mmol/L(參 考 值:3.5~5.5 mmol/L),雄烯二酮<1.05 nmol/L,脫氫表雄酮<0.41 mmol/L,卵泡刺 激 素83.30 mIU/mL(參 考 值:2.8~11.3 mIU/mL ),促黃體生成素34.80 mIU/mL(參考值:1.1~11.6 mIU/mL),泌乳素267 mIU/L(參考值:40~530 mIU/L),孕酮44.20 nmol/L(參考值:0.64~3.60 nmol/L),雌二醇75.60 pmol/L(參考值:73.4~587.0 pmol/L),睪酮<0.69 nmol/L。醛固酮(aldosterone,ALd)臥立位實驗:臥位ALd 0.09 ng/mL(參考值:0.06~0.17 ng/mL),血漿腎素活性(plasma renin activity,PRA)0.1 ng/mL(參考值:0.50~0.79 ng/mL),血管緊張素Ⅱ(angiotensin-Ⅱ,AT-Ⅱ)32.0 ng/mL(參 考 值:28.2~52.3 ng/mL),立位ALd 0.13 ng/mL(參考值:0.07~0.30 ng/mL),PRA 0.08 ng/mL(參 考 值:0.93~6.56 ng/mL),AT-Ⅱ 32.0 ng/mL(參考值:55.3~115.3 ng/mL)。促腎上腺皮質激 素-皮 質 醇(adrenocorticotropic hormone-cortisol,ACTH-COR)節律如下,ACTH 8:00為 81.34 pg/mL,15:00為39.82 pg/mL,24:00為22.97 pg/mL(參考值:7.2~63.3 pg/mL);COR 8:00為31.9 nmol/L,15:00為73.38 nmol/L,24:00為14.61 nmol/L(參考值:上午64~327 nmol/L,下午171~536 nmol/L)。甲功化驗結果:FT3略升高,FT4、TSH均正常。
腎上腺增強CT顯示左腎上腺略增粗。婦科超聲檢查顯示,子宮內膜顯示不清,雙側卵巢內均未見卵泡,子宮發育不良。腕關節平片顯示,骨骼發育延遲,骨化中心7枚骨化核,骨齡9歲。染色體分析:46,XX,inv(9)(p11q13)。垂體磁共振檢查發現疑似垂體增生,右側大腦中動脈狹窄。顱腦磁共振血管成像(magnetic resonance angiography,MRA)檢查發現,雙側大腦動脈改變,提示煙霧病。
1.3 基因檢測
在CYP17A1基因編碼區,檢出c.287G>T 雜合突變(R96L)突變及c.1117delC 雜合突變(表1)。經Mutation taster軟件預測為有害突變,但尚未發現有相關的文獻報道。
1.4 診斷

表1 先天性腎上腺皮質增生癥基因檢測報告
CAH(17α-OHD);煙霧病可能性大。
1.5 治療方案
給予醋酸潑尼松早2.5 mg、晚2.5 mg口服,鈣爾奇D 600 mg/d口服。住院期間補鉀后血鉀升至正常,由于患者血壓高,給予硝苯地平片口服(10 mg,2次/d),具體治療情況見表2。
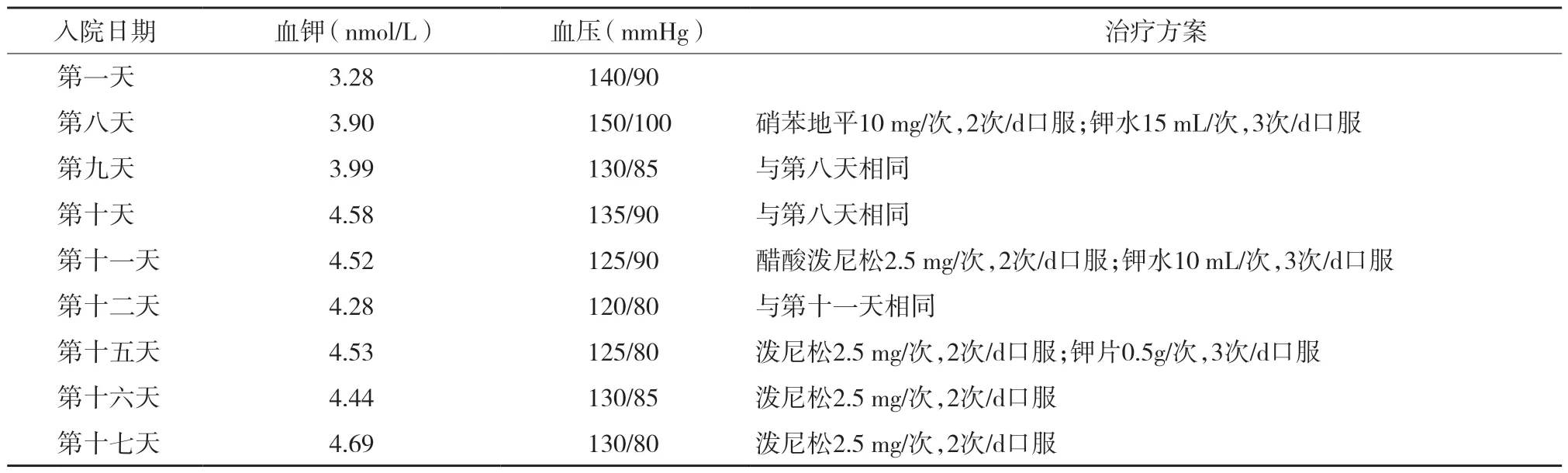
表2 住院期間診治情況
1.6 隨訪
患者于2012年12月11日出院,出院后在門診進行隨訪,血鉀水平正常,血壓較前有所下降,波動于105~125/60~80 mmHg(表3)。因患者患有煙霧病,腦出血及腦梗死風險大,因此建議其至神經外科行手術治療。
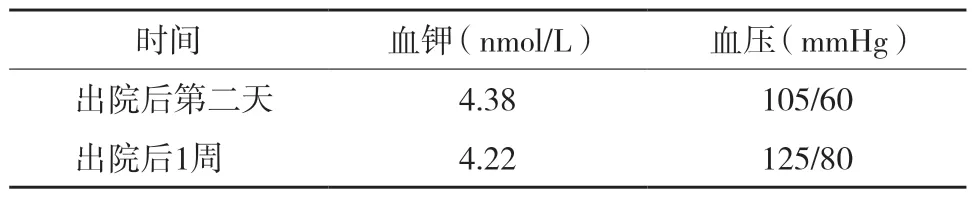
表3 出院后隨診情況
2 討論
CAH是一種罕見的常染色體隱性遺傳的內分泌疾病。由于與皮質類固醇合成有關的酶活性先天缺乏,皮質醇的合成被部分或完全阻斷[1],從而增加了下丘腦-垂體中促腎上腺皮質激素釋放激素-促腎上腺皮質激素的代償性分泌,并導致腎上腺皮質增生、雄激素和一些中間代謝產物增加[2-3]。CAH種類繁多,新生兒疾病的發病率從1∶10 000到1∶20 000,具有明顯的種族差異,女性多于男性。本例CAH屬于17α-OHD,相對罕見,約占CAH的1%[4]。
編碼17α-羥化酶的基因CYP17A1位于人類常染色體10q24225,編碼508個氨基酸的蛋白,即17α-羥化酶/17,20-碳鏈裂解酶,該酶兼有 17α-羥化酶和 17,20-裂解酶2種活性,前者催化孕烯醇酮和孕酮轉變為 17α-羥孕烯醇酮和17α-羥孕酮,后者使 17,20 位碳鏈裂解,形成雌激素的前體,即脫氫表雄酮和雄烯二酮;在腎上腺和生殖腺中表達[5]。
本例患者檢測出CYP17A1基因的c.287G>T 雜合突變(R96L)突變及c.1117delC 雜合突變,屬于臨床意義未明突變,分別導致了由精氨酸(堿性氨基酸)轉變為亮氨酸(非極性氨基酸)和蛋白長度的縮短和序列的改變,由509AA縮短至418AA,且372~418AA序列發生改變。經 SIFT、polyphen-2、mutation taster 軟件預測均為有害突變,但在 dbSNP、千人基因組、hapmap 數據庫中均無頻率,文獻中也無 R96L 突變位點的相關報道。先天性腎上腺皮質增生還存在其他的致病基因,而本次檢測只包含CYP11B1,CYP11B2,CYP17A1,CYP21A2基 因,不排除其他基因存在致病位點的可能性。這種突變可能導致患者的17α-羥化酶合成缺乏和活性下降,17-羥孕烯醇酮及17-羥孕酮合成受阻,脫氧皮質醇及雄烯二酮水平下降,導致糖皮質激素及性激素缺乏;同時,17-羥孕烯醇酮及17-羥孕酮的前體相應增多,具有鹽皮質激素作用的去氧皮質酮產生增多,使PRA和ALD的生成也受到抑制[6],主要表現為疲勞、厭食、惡心、嘔吐、皮膚及黏膜色素沉著、低血鉀、血ACTH升高和不同程度高血壓。男性患兒(46,XY)可表現為女性化,外生殖器為女性或部分男性,無子宮和輸卵管,隱睪,睪丸可位于腹股溝或腹腔,骨齡落后。女性患者(46,XX)以性發育不成熟、原發閉經、腋毛、陰毛缺乏、陰部年輕、體型高大、骨齡下降為特征。少數患兒性發育可正常。
絕大多數CAH患者都需要糖皮質激素治療,但在使用中應注意劑量,在保持患者的正常生長發育和生活質量的同時,又應避免醫源性類庫欣綜合征的出現。對于未成年患者以氫化可的松治療為首選,也可選用強的松,成年患者首選地塞米松。因為此類患者不能合成足夠的性激素,所以其性發育并不能在單純糖皮質激素治療后恢復正常,絕大多數患者還可選擇性激素及手術治療。應根據染色體的性別、性腺的性別、外生殖器的分化、發育等進行鑒別。染色體為46,XX的患者,在16~18歲開始可給雌激素和孕激素給予人工循環治療。染色體為46,XY的患者可以預防性切除隱睪,按社會性別及個人選擇,決定是否補充雌激素治療。本例患者未成年,選擇給予醋酸潑尼松(強的松)早、晚各口服2.5 mg。待煙霧病得到有效治療后,還應給予患者人工月經治療。
