Dubin-Johnson綜合征分子遺傳學研究進展
2019-12-06 05:56:32張紅艷李亞絨張艷敏車鳳玉
中國婦幼健康研究 2019年11期
張紅艷,李亞絨,張艷敏,車鳳玉
(1.西安醫學院,陜西 西安 710021;2.西安市兒童醫院,陜西 西安 710003)
Dubin-Johnson綜合征(Dubin-Johnson syndrome,DJS)由Dubin和Johnson于1954年首次發現并報道[1]。DJS是一種罕見的常染色體隱性遺傳病,目前報道西帕迪奇猶太人群中發病率較高,約1∶3 000[2]。ATP結合盒C亞家族成員ABCC2基因突變致多藥耐藥相關蛋白(multidrug resistance associated proteins,MRP2)功能障礙或缺失是DJS發病的重要機制。臨床以持續性或間歇性皮膚、鞏膜及小便發黃(輕至中度)為主要表現,可有右上腹不適或隱痛、乏力、食欲不振、惡心、嘔吐等表現,約半數患者可有輕度肝臟腫大或輕微肝區壓痛,脾腫大者少見,無皮膚瘙癢[3]。DJS患者肝臟外觀呈黑色,組織學表現為肝小葉中央區肝細胞溶酶體內有棕褐色色素顆粒沉著,肝臟病理活檢為確診的金準標。DJS臨床癥狀一般比較輕微,預后良好,一旦確診無需特殊治療,口服熊去氧膽酸和苯巴比妥鈉對癥即可[1]。本文就DJS分子遺傳學研究進展進行綜述。
1 MRP2的結構
MRP2是一種190kDa的完整膜糖蛋白,含有1 545個氨基酸,它包含兩個ATP結合磁帶和17個跨膜序列,主要分布于肝細胞的極化上皮根尖小管膜上,以及其他具有極化細胞的頂膜上,如腸細胞和腎小管細胞,是一種ATP依賴的兩親陰離子輸出泵,可用于共軛和非共軛兩親陰離子的輸出。MRP2的共軛輸出泵及其特異性小管異構體,使其底物很廣泛,包括非膽汁酸有機陰離子轉運體、多特異性有機陰離子轉運體、谷胱甘肽S共軛輸出泵及白三烯輸出泵[4-5]。MRP2功能障礙或缺失,使得肝細胞中結合膽紅素及其他有機陰離子向毛細膽管排泄障礙,引起膽紅素在體內淤積,從而導致慢性的、以結合膽紅素升高為主的高膽紅素血癥[6]。
2 MRP2的編碼基因ABCC2及其突變
MRP2的編碼基因ABCC2(ATP結合盒C亞家族成員)于1997年被Paulusma等在高膽紅素血癥大鼠中發現,同年谷口一等在順鉑耐藥腫瘤細胞中發現了人類ABCC2基因,該基因位于10號染色體短臂24帶(10q24),1999年Tsujii等[7]發現該基因由32個外顯子組成。迄今為止已報導的DJS相關的ABCC2基因突變位點多達68個,包括:錯義突變、無義突變、剪接突變、調節突變、刪除/缺失突變、插入突變、小插入-缺失突變、復雜重排突變,其中絕大多數是堿基置換突變導致的錯義突變和無義突變,見表1。
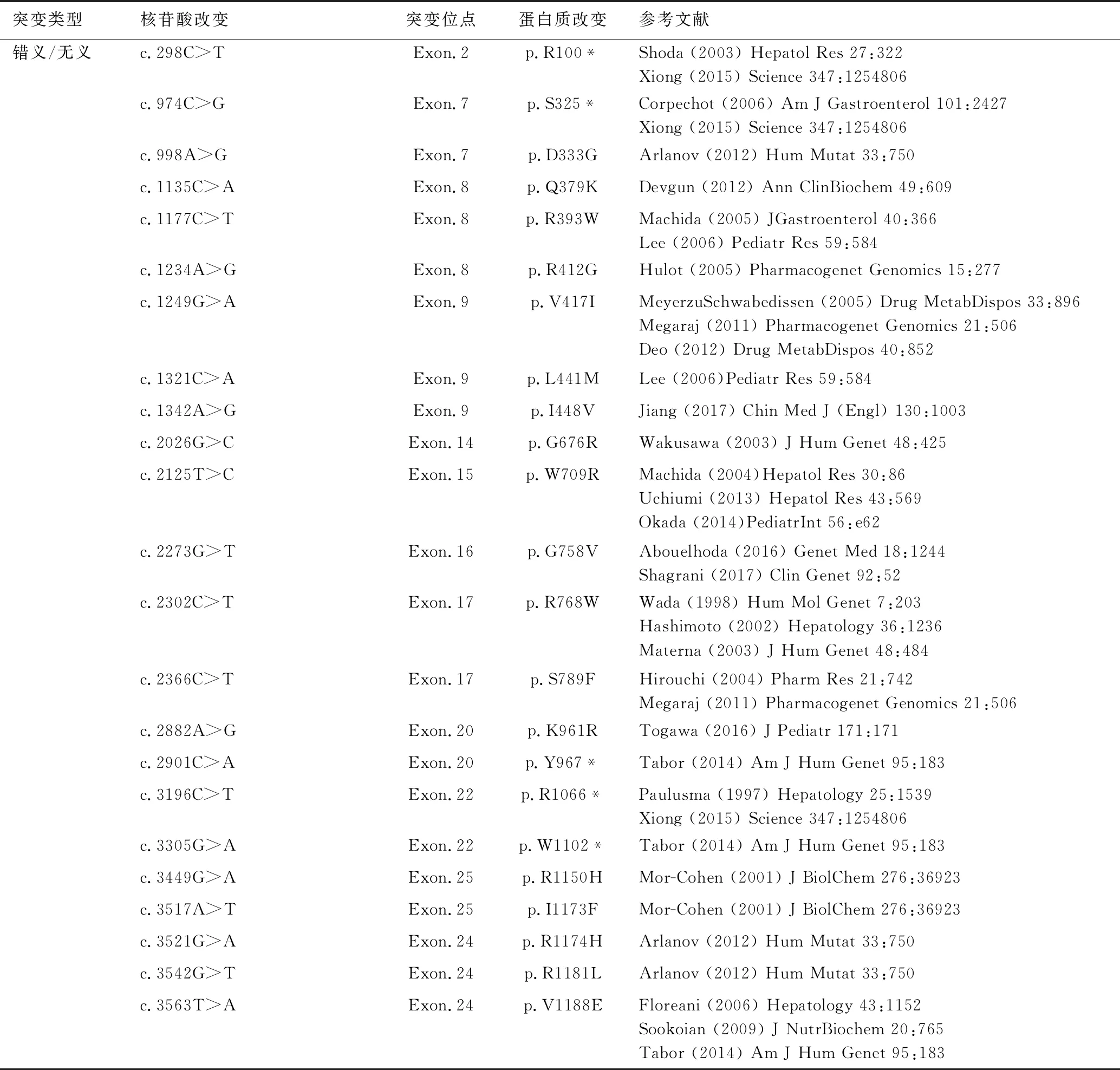
表1 Dubin-Johnson綜合征編碼基因ABCC2突變類型
續上表
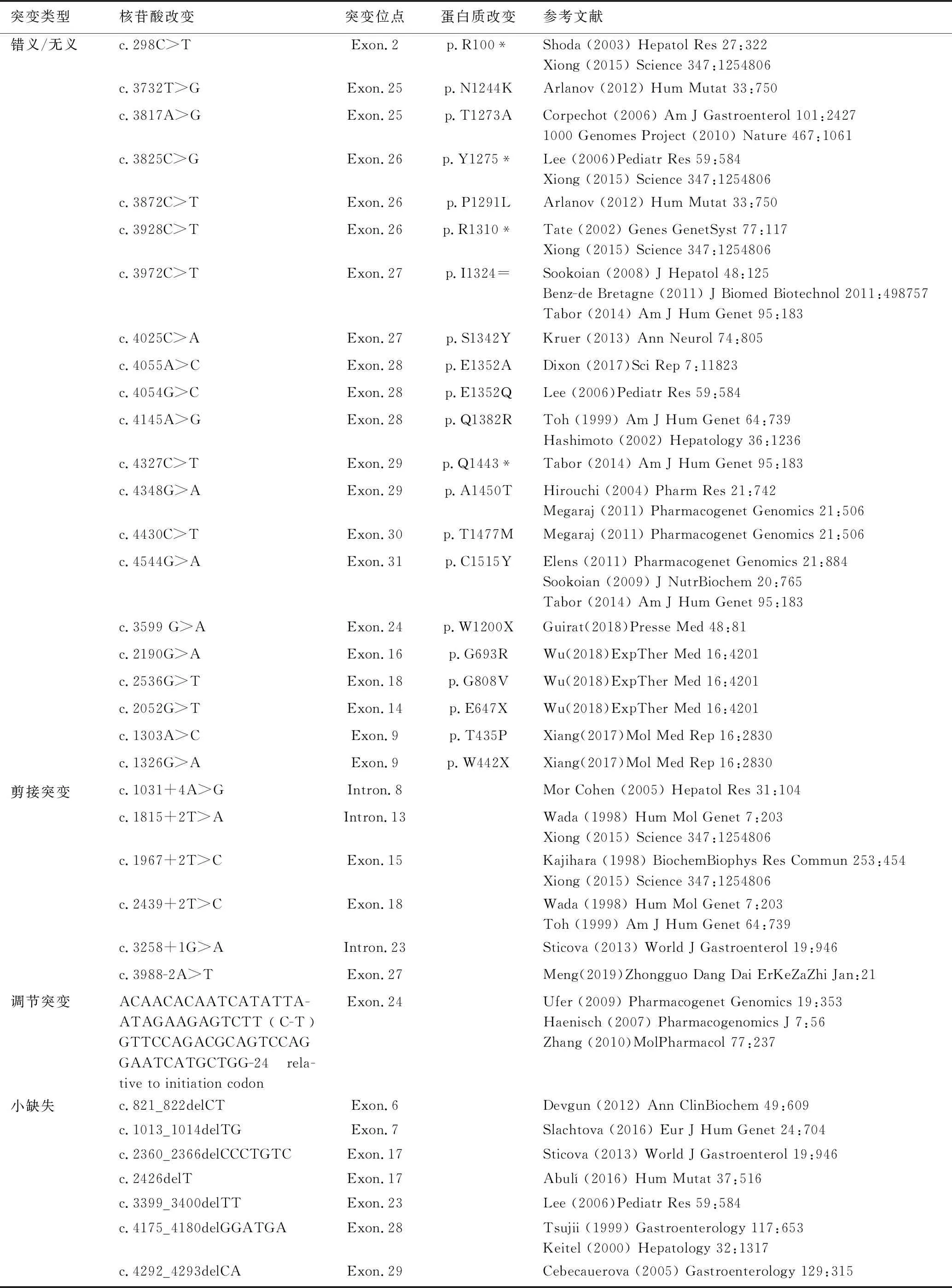
突變類型核苷酸改變突變位點蛋白質改變參考文獻錯義/無義c.298C>TExon.2p.R100*Shoda(2003)HepatolRes27:322Xiong(2015)Science347:1254806c.3732T>GExon.25p.N1244KArlanov(2012)HumMutat33:750c.3817A>GExon.25p.T1273ACorpechot(2006)AmJGastroenterol101:24271000GenomesProject(2010)Nature467:1061c.3825C>GExon.26p.Y1275*Lee(2006)PediatrRes59:584Xiong(2015)Science347:1254806c.3872C>TExon.26p.P1291LArlanov(2012)HumMutat33:750c.3928C>TExon.26p.R1310*Tate(2002)GenesGenetSyst77:117Xiong(2015)Science347:1254806c.3972C>TExon.27p.I1324=Sookoian(2008)JHepatol48:125Benz-deBretagne(2011)JBiomedBiotechnol2011:498757Tabor(2014)AmJHumGenet95:183c.4025C>AExon.27p.S1342YKruer(2013)AnnNeurol74:805c.4055A>CExon.28p.E1352ADixon(2017)SciRep7:11823c.4054G>CExon.28p.E1352QLee(2006)PediatrRes59:584c.4145A>GExon.28p.Q1382RToh(1999)AmJHumGenet64:739Hashimoto(2002)Hepatology36:1236c.4327C>TExon.29p.Q1443*Tabor(2014)AmJHumGenet95:183c.4348G>AExon.29p.A1450THirouchi(2004)PharmRes21:742Megaraj(2011)PharmacogenetGenomics21:506c.4430C>TExon.30p.T1477MMegaraj(2011)PharmacogenetGenomics21:506c.4544G>AExon.31p.C1515YElens(2011)PharmacogenetGenomics21:884Sookoian(2009)JNutrBiochem20:765Tabor(2014)AmJHumGenet95:183c.3599G>AExon.24p.W1200XGuirat(2018)PresseMed48:81c.2190G>AExon.16p.G693RWu(2018)ExpTherMed16:4201c.2536G>TExon.18p.G808VWu(2018)ExpTherMed16:4201c.2052G>TExon.14p.E647XWu(2018)ExpTherMed16:4201c.1303A>CExon.9p.T435PXiang(2017)MolMedRep16:2830c.1326G>AExon.9p.W442XXiang(2017)MolMedRep16:2830剪接突變c.1031+4A>GIntron.8MorCohen(2005)HepatolRes31:104c.1815+2T>AIntron.13Wada(1998)HumMolGenet7:203Xiong(2015)Science347:1254806c.1967+2T>CExon.15Kajihara(1998)BiochemBiophysResCommun253:454Xiong(2015)Science347:1254806c.2439+2T>CExon.18Wada(1998)HumMolGenet7:203Toh(1999)AmJHumGenet64:739c.3258+1G>AIntron.23Sticova(2013)WorldJGastroenterol19:946c.3988-2A>TExon.27Meng(2019)ZhongguoDangDaiErKeZaZhiJan:21調節突變ACAACACAATCATATTA-ATAGAAGAGTCTT(C-T)GTTCCAGACGCAGTCCAGGAATCATGCTGG-24rela-tivetoinitiationcodonExon.24Ufer(2009)PharmacogenetGenomics19:353Haenisch(2007)PharmacogenomicsJ7:56Zhang(2010)MolPharmacol77:237小缺失c.821_822delCTExon.6Devgun(2012)AnnClinBiochem49:609c.1013_1014delTGExon.7Slachtova(2016)EurJHumGenet24:704c.2360_2366delCCCTGTCExon.17Sticova(2013)WorldJGastroenterol19:946c.2426delTExon.17Abulí(2016)HumMutat37:516c.3399_3400delTTExon.23Lee(2006)PediatrRes59:584c.4175_4180delGGATGAExon.28Tsujii(1999)Gastroenterology117:653Keitel(2000)Hepatology32:1317c.4292_4293delCAExon.29Cebecauerova(2005)Gastroenterology129:315
續上表
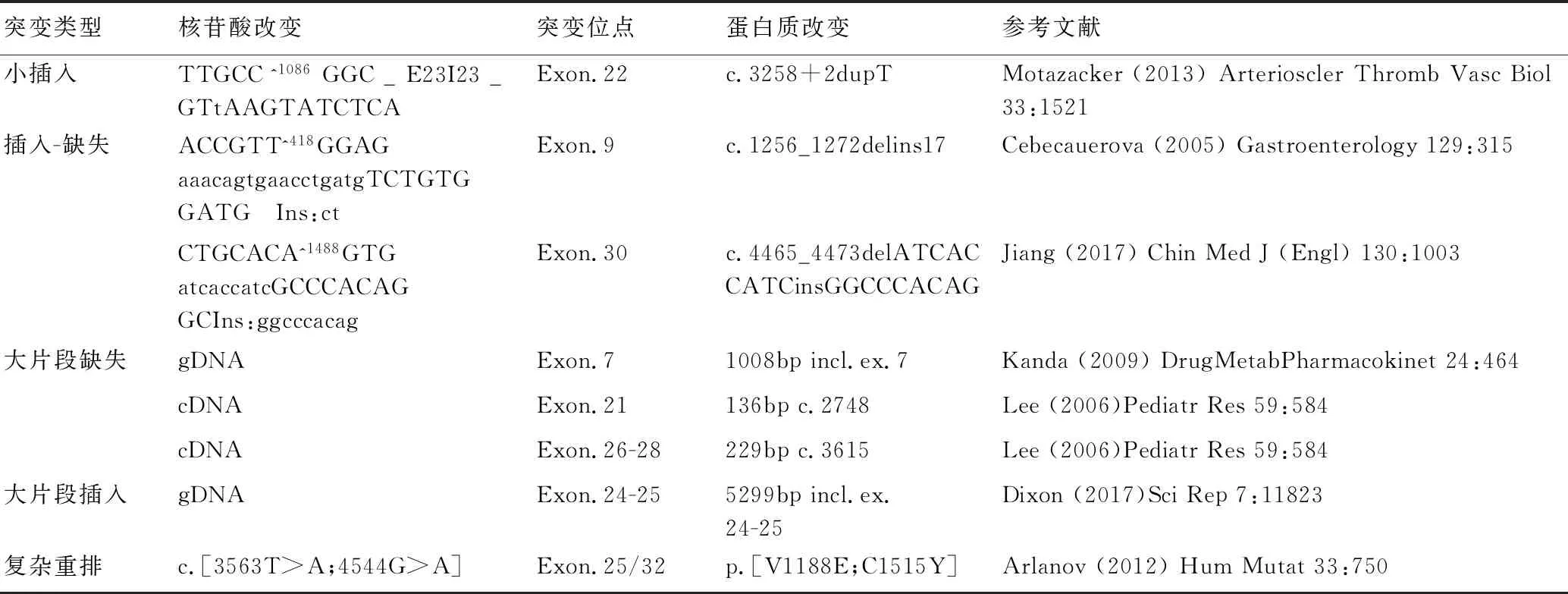
突變類型核苷酸改變突變位點蛋白質改變參考文獻小插入TTGCC^1086GGC_E23I23_GTtAAGTATCTCAExon.22c.3258+2dupTMotazacker(2013)ArteriosclerThrombVascBiol33:1521插入-缺失ACCGTT^418GGAGaaacagtgaacctgatgTCTGTGGATG Ins:ctExon.9c.1256_1272delins17Cebecauerova(2005)Gastroenterology129:315CTGCACA^1488GTGatcaccatcGCCCACAGGCIns:ggcccacagExon.30c.4465_4473delATCACCATCinsGGCCCACAGJiang(2017)ChinMedJ(Engl)130:1003大片段缺失gDNAExon.71008bpincl.ex.7Kanda(2009)DrugMetabPharmacokinet24:464cDNAExon.21136bpc.2748Lee(2006)PediatrRes59:584cDNAExon.26-28229bpc.3615Lee(2006)PediatrRes59:584大片段插入gDNAExon.24-255299bpincl.ex.24-25Dixon(2017)SciRep7:11823復雜重排c.[3563T>A;4544G>A]Exon.25/32p.[V1188E;C1515Y]Arlanov(2012)HumMutat33:750
注:*代表翻譯提前終止;=代表氨基酸變化相同。
2.1錯義突變和無義突變
2018年Togawa等[8]招募了10名新生兒期出現膽汁淤積,最終診斷為DJS的患兒,其中9例日本患兒,1例中國患兒。8例患兒在新生兒期進行了肝組織穿刺活檢術,3例為黑肝,6例經病理免疫組化研究證實缺乏MRP2表達。所有患兒均進行基因檢測,共發現ABCC2基因11個致病性突變位點,其中c.2882 C>T,p.K961R為新的致病性突變,認為錯義突變和無義突變共同阻止了MRP2在肝小管膜中的表達,從而可能導致DJS。
2017年Xiang等[9]對1例臨床診斷DJS的患者進行基因測序,在ABCC2基因的第10外顯子發現了2個新突變,即錯義突變:c.1303A>C,p.T435P,遺傳自母親,無義突變c.1326G>A,p.W442X,遺傳自父親,符合DJS常染色體隱性遺傳。對突變后MRP2蛋白質功能研究,證實了錯義突變c.1303A>C,p.T435P和無義突變c.1326G>A,p.W442X破壞了MRP2蛋白質的功能,從而導致DJS,由此證實了該患者DJS的臨床診斷。
2019年Guirat等[10]對1名表現為黃疸的9歲男童進行ABCC2基因測序,在第25外顯子發現一個新的致病突變,即無義突變:c.3599 G>A,p.W1200X,為純合突變,遺傳來自父母,從而診斷該患兒為DJS。本例患兒未進行肝組織穿刺活檢。此外2014年Tabor等發現:c.3305 G>A,p.W1102*等新的致病性突變陸續被報道,至今已報道的錯義突變和無義突變共有42個。
2.2剪接突變
2013年Sticova等[11]對一名癌癥患者的隨訪中發現該患者表現為持續顯著的高膽紅素血癥,肝組織穿刺活檢、病理免疫組化研究證實了MRP2表達正常,基因測序確定了ABCC2基因的兩個新致病性突變:即缺失突變:c.2360_2366delCCCTGTC和剪接突變:c.3258 + 1G>A,前者位于第18外顯子,為雜合缺失;后者位于第23內含子,該突變在第23個剪接供體處發生了G>A的替換,上述位點的突變改變了RNA前體正常的剪接方式,引起mRNA的異常剪接,由此確診該患者為DJS。2019年Meng等[12]通過基因檢測確診了1例DJS患兒,發現了ABCC2基因1個新的致病性突變:c.3988-2A>T,遺傳自母親。此類突變最早由Kajihara在1998年發現:c.1967+2T>C,隨后相繼發現 c.1815+2T>A,c.2439+2T>C和 c.1031+4A>G共6個剪接突變。
2.3缺失/刪除突變
1999年Tsujii等[7]對2例DJS患者進行研究,檢測了ABCC2基因32個外顯子,以及外顯子-內含子的邊界區域,除了發現已報道的無義突變:c.1066 C>T,同時在2例患者發現了第1 392到1 394核苷酸之間6個核苷酸的缺失突變,致使其下游的開放閱讀框架發生了改變,最終導致精氨酸和蛋氨酸的缺失,患者的肝細胞小管膜MRP2不表達。
2016年Slachtova等[13]對7個羅姆人家系56名成員進行研究,共發現17例DJS患者和30例雜合子攜帶者。該研究在ABCC2基因的第8外顯子發現了一個新的缺失突變c.1013_1014 delTG,17例DJS患者均為純合子的c.1013_1014 delTG,導致被編碼的MRP2發生移碼和截短,由原來的1545個氨基酸縮短為352個氨基酸。研究認為ABCC2基因的c.1013_1014 delTG突變可能導致MRP2的mRNA快速降解,從而導致MRP2的合成、穩定性及轉運方面的缺陷。研究在7個無相關性的羅姆人家系中發現相同的c.1013_1014 delTG變異,推斷c.1013_1014 delTG變異為羅姆人的熱點變異。2016年Slachtova等[13]發現的ABCC2基因新的致病性缺失突變:c.2360_2366delCCCTGTC,該突變位于第18外顯子,為第2 360到2 366核苷酸之間7個核苷酸的缺失突變,使其下游的開放閱讀框架發生了改變,最終導致氨基酸的缺失,從而影響MRP2的功能。
2006年Lee等[14]對4例肝組織病理符合國內DJS診斷標準的患者進行研究,其中2名患者新生兒期即出現膽汁淤積并診斷,另外2例有新生兒黃疸和高膽紅素血癥史,于青春期診斷DJS,并對4例患者進行長達5~20年隨訪。在4例患者中共發現ABCC2基因的6個突變位點,其中5個突變位點為已報道的,包括缺失突變2748del136和3615del22,錯義突變p.L441M和p.E1352Q以及無義突變p.Y1275*,此外還發現一個未經報道的雜合子的缺失突變:c.3399_3400delTT,該突變使得42個氨基酸移位及終止密碼子提前出現,致使蛋白質合成中斷,最終導致ABC結構域的丟失。該研究表明,MRP2中具有重要功能的ABC結構域的丟失可能與DJS的早期發病相關。
2009年Kanda等[15]對1例臨床診斷為DJS患者基因測序,發現了ABCC2基因一個新的突變位點:gDNA 1008 bpincl.Ex.7,該突變為純合的1008 bp的外顯子7大片段缺失,患者未進行肝組織穿刺活檢,無法確定外顯子7的1 008 bp大規模缺失對MRP2功能的影響,推測該缺失影響MRP2合成。
迄今為止共發現的缺失/刪除突變14個,其中11個小缺失、3個大片段缺失。
2.4小插入-缺失突變
2005年Cebecauerova等[16]報道一例3歲男孩攜帶ABCC2基因2個新的突變:其中一個為外顯子10的雜合插入-缺失:indel 1256 insCT/delAAACAGTGAACCTGATG,遺傳自父親,另一個為外顯子30的雜合缺失4292delCA,遺傳自母親,后者的突變導致了氨基酸的移位和1 461位提前出現了終止密碼子。但患者肝臟并不表現為黑肝,且組織學正常,由此得出結論:對于診斷不明確的、懷疑有遺傳性高膽紅素血癥的患者,無論其表型如何,無創的DNA測序分析應為首選方法,當基因測序不能明確時,才應選取有創的肝組織穿刺活檢檢查。
2017年Jiang等[17]對一名血清膽紅素持續輕度升高的女性患者進行研究,發現了ABCC2基因新的突變c.4712_4720delinsGGCCCACAG(父源)和p.I448V(母源),兩者均被證實是致病性突變,上述突變通過降低蛋白質的穩定性,嚴重破壞轉運蛋白的活性,從而可能導致DJS。
2.5插入突變
2017年Dixon等[18]在對妊娠肝內膽汁淤積癥患者的研究中,發現了ABCC2基因的一個新突變:gDNA Dup 5299 bp incl.ex.24-25,該突變是包括外顯子24和25在內的5 299個堿基對的基因組重復,該研究預測這個突變會導致一個早熟的終止密碼子提前插入,使得其下游的閱讀框發生改變,使多肽鏈的氨基酸種類和序列發生改變。同時還發現一個已報道的錯義突變,c.4055 A>C,可能導致p.Glu1352Ala的改變,為有害突變。迄今為止發現此類突變有2個,其中小插入1個、大片段插入1個。
3總結
ABCC 2基因的分子遺傳學分析對DJS的診斷具有重要意義。通過基因檢測可明確患者是否患有DJS,從而減少患者反復不必要的就診,減少不必要的檢查及治療,同時還可減少肝組織穿刺活檢等有創侵入性操作。近年來通過基因檢測手段確診的DJS患者逐年增加,新的致病性突變不斷被發現,豐富了人類基因組突變數據庫,使得人們對DJS的認識也更加全面。
