高效液相色譜串聯質譜法檢測馬鈴薯及馬鈴薯粉中α-茄堿和α-卡茄堿含量
2019-11-27 11:28:30,3,3,3,*
食品工業科技 2019年21期
關鍵詞:優化
,3,3,3,*
(1.農業部農產品質量監督檢驗測試中心(北京),北京 100083;2.農業農村部科技發展中心,北京 100045;3.中國農業大學食品科學與營養工程學院,北京 100083)
馬鈴薯(Solanumtuberosum)是茄科多年生草本植物,在世界范圍內廣泛種植[1]。由于其耐干旱,穩產高產,抗災力強,成為世界上僅次于小麥、水稻、玉米的第四大糧食作物[2],距今已有7000多年的栽培歷史[3]。由于馬鈴薯營養均衡、產業鏈長等特點,被廣泛應用于主食消費和加工利用,是歐美、非洲等70多個國家的主食作物[4]。
茄堿是存在于馬鈴薯中的天然的甾體糖苷類生物堿毒素,又名龍葵堿、龍葵素,主要由葡萄糖殘基和茄啶組成的一種弱堿性糖苷[5]。馬鈴薯中的茄堿主要是α-茄堿(α-solanine)和α-卡茄堿(α-chaconine)[6],其含量占糖苷類生物堿總量的90%以上[7]。在馬鈴薯的芽、未成熟塊莖、成熟薯皮及靠近薯皮的組織中含量較高[8]。茄堿通過抑制膽堿酯酶活性而對胃腸道黏膜有較強的刺激性和腐蝕性[9]、對中樞神經有麻痹作用、對紅細胞有溶血作用[10],可引起急性腦水腫、胃腸炎等。中毒的主要表現為胃痛加劇,惡心嘔吐,呼吸困難,伴隨全身虛弱和衰竭[11],也可以致畸[12],導致腦畸形和脊柱裂[13],嚴重可導致死亡[14]。由此可見,茄堿中毒是不容忽視的食品安全問題[15]。
目前茄堿提取方法主要有勻漿提取法[16]、超聲提取法[17]、微波輔助提取法[18]、攪拌浸提法[19]、回流提取法等[20],檢測方法多采用高效液相色譜[21-24],液相色譜串聯質譜聯用法[25-28]。傳統的提取方法多采用酸提堿沉的方法,時間長、步驟復雜,回收率偏低,而用液相色譜方法時,檢測波長在紫外區,易受到雜質干擾,難以用于較復雜樣品的檢測。此外,馬鈴薯中茄堿的檢測方法較為陳舊,我國現有的茄堿檢測方法不完善,因此,如何改進馬鈴薯中茄堿的提取條件、提高回收率,建立新的檢測技術,已成為該研究領域急待解決的問題[29]。
本文通過乙酸水溶液對馬鈴薯及馬鈴薯粉中的α-茄堿和α-卡茄堿進行提取,馬鈴薯樣品提取后直接過膜上機,馬鈴薯粉采用C18 SPE柱凈化后即可過膜上機,操作簡便、高效、快速,避免了超聲、振蕩、回流和離心等耗時長的前處理方式;采用高效液相色譜-串聯質譜法檢測,重復性好、精密度好、檢測效率高,不但可以避免了雜質干擾的影響,還可以完成α-茄堿和α-卡茄堿的同時檢測,為低含量茄堿的檢測提供了準確可靠的實用方法。
1 材料與方法
1.1 材料與儀器
馬鈴薯及馬鈴薯粉 購自超市,馬鈴薯編號為S1~S12,馬鈴薯粉編號為S13~S15。甲醇、乙腈、無水乙醇、甲酸和冰乙酸 色譜純;甲酸銨 分析純;α-茄堿(20562-02-1,純度≥99%)和α-卡茄堿(20562-03-2,純度≥99%) 北京百靈威科技有限公司;Milli Q超純水。
Agilent 1260/6460高效液相色譜-串聯質譜儀 安捷倫科技有限公司;T25高速勻漿機 北京德泉興業商貿有限公司;RE-52AA旋轉蒸發儀 上海振捷實驗設備有限公司;SHB-III循環水式多用真空泵 鄭州長城科工貿有限公司;C18固相萃取柱柱(500 mg/3 mL) 安捷倫科技有限公司。
1.2 實驗方法
1.2.1 質譜條件優化 采用直接進樣的方式,分別在正離子和負離子模式下對α-茄堿和α-卡茄堿的標準溶液進行掃描,優化質譜條件。
1.2.2 色譜條件優化 實驗比較了0.1%甲酸-乙腈,2 mmol/L甲酸銨(含0.1%甲酸)-乙腈、5 mmol/L甲酸銨(含0.1%甲酸)-乙腈,10 mmol/L甲酸銨(0.1%甲酸)等流動相體系對其分離效果的影響。
1.2.3 馬鈴薯中茄堿的提取方法 馬鈴薯去掉表面泥土,切塊后制成勻漿,稱取5.0 g勻漿后的樣品于200 mL燒杯中,加入不同比例的提取溶劑,采用不同的方法進行提取,提取液定容至100 mL,上機前取2 mL提取液用流動相定容至10 mL,過0.22 μm微孔濾膜后分析。
1.2.3.1 提取溶劑優化 分別用乙酸-水-NaHSO3(5/100/0.5,v/v/w)、5%乙酸水溶液、10%乙酸水溶液、15%乙酸水溶液、1%乙酸乙醇溶液、5%乙酸乙醇溶液和10%乙酸乙醇溶液等7種溶液作為提取劑對馬鈴薯中茄堿進行提取,比較不同提取溶劑的提取效果。
1.2.3.2 提取方式的優化 分別采用勻漿提取[16]、超聲提取[17]、回流提取[20]及振蕩提取[27]方式對馬鈴薯中的茄堿進行提取,比較不同提取方式的提取效果。
1.2.3.3 料液比的優化 采用勻漿提取方式,以1∶2、1∶3、1∶5、1∶10等不同料液比對馬鈴薯中茄堿提取效果的影響。
1.2.3.4 提取時間的優化 以10%的乙酸水溶液為提取液,料液比為1∶10,采用勻漿提取方式,比較提取為1、2、3、4 min的提取效果。
1.2.4 馬鈴薯粉中茄堿的提取方法 稱取1.0 g馬鈴薯粉于200 mL燒杯中,加10 mL的10%乙酸水浸泡30 min,再加40 mL的10%乙酸水,勻漿提取3 min,轉移至100 mL容量瓶中,用10%乙酸水定容。
1.2.5 凈化條件的優化 取馬鈴薯粉提取液30 mL于離心管中,10000 r/min離心5 min。取上清液5 mL加到預先用5 mL甲醇,5 mL 10%乙酸水溶液活化的固相萃取柱中,5 mL 15%甲醇水溶液淋洗,分別用不同洗脫液進行洗脫。取1 mL洗脫液用流動相稀釋10倍后,過0.22 μm微孔濾膜后進行分析。
1.2.6 色譜條件
1.2.6.1 液相色譜條件 色譜柱:Agilent C18柱(100 mm×3.0 mm,2.7 μm);流動相A為5 mmol/L甲酸銨水溶液(含0.1%甲酸),流動相B為乙腈;梯度洗脫條件:78%的A保持2 min,5 min后變為65%的A保持2 min;柱溫30 ℃;進樣量3 μL;流速0.30 mL/min。
1.2.6.2 質譜條件 毛細管電壓:3.5 kv;噴嘴電壓:500 V;干燥氣體溫度:350 ℃;干燥氣流量:10 L/min;鞘氣溫度:300 ℃;鞘氣流速:11 L/min。
1.2.7 標準溶液配制 分別稱取α-茄堿和α-卡茄堿標準品10.0 mg,加甲醇溶解并定容至10.0 mL,配成濃度為1.0 mg/mL的標準儲備液。吸取α-茄堿和α-卡茄堿標準儲備液,用甲醇逐級稀釋,配制成濃度分別為0.010、0.025、0.050、0.10、0.25、0.50和1.00 μg/mL的混合標準工作液。
1.2.8 樣品中茄堿含量計算
其中,X為試樣中待測組分含量,單位為毫克每千克(mg/kg);ρ為標準曲線得出的試樣中α-茄堿或α-卡茄堿的質量濃度,單位為微克每毫升(μg/mL);V為試樣定容體積,單位為毫升(mL);m為試樣的質量,單位為克(g);f為稀釋倍數;1000為單位換算系數。
1.2.9 方法的準確度和精密度 分別按1.2.3和1.2.4稱取一定量的馬鈴薯及馬鈴薯粉樣品于燒杯中,添加一定濃度的α-茄堿和α-卡茄堿的標準溶液,使馬鈴薯中α-茄堿和α-卡茄堿添加水平分別為5.00、20.0和50.0 mg/kg;馬鈴薯粉中α-茄堿和α-卡茄堿的添加水平分別為50.0、100和200 mg/kg,放置30 min后,按所建立的方法進行提取測定,計算樣品的添加回收率。
1.3 數據處理
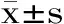
2 結果與分析
2.1 質譜條件的優化
分別在正離子和負離子模式下對α-茄堿和α-卡茄堿進行母離子掃描,設置掃描范圍為300~900 amu,結果表明,兩種茄堿在正離子模式下響應較高。確定掃描方式、母離子后,優化碎裂電壓、碰撞能量,優化后的多反應監測參數見表1。
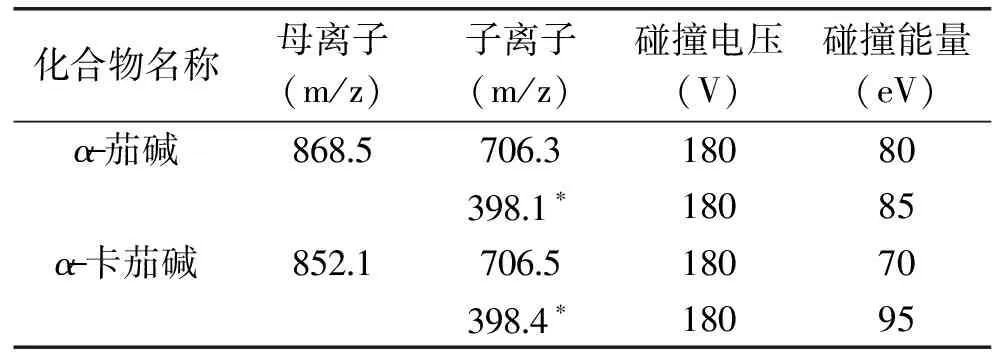
表1 α-茄堿和α-卡茄堿多反應監測參數Table 1 Parameters of MRM for α-solanine and α-chaconine
注:*為定量離子。
2.2 流動相的優化
α-茄堿和α-卡茄堿結構類似,在色譜中的保留時間相近,為了達到較好的分離效果,提高離子化效率,實驗對流動相組成及洗脫條件進行了優化。結果表明,流動相中加入甲酸銨后α-茄堿和α-卡茄堿分離度增加。隨著甲酸銨濃度的增加,α-茄堿的響應值有所提高而α-卡茄堿的響應值有所下降。綜合考慮響應值和分離度的影響,實驗選擇5 mmol/L甲酸銨(含0.1%甲酸)-乙腈作為流動相。α-茄堿和α-卡茄堿標準溶液總離子及提取離子色譜圖見圖1。
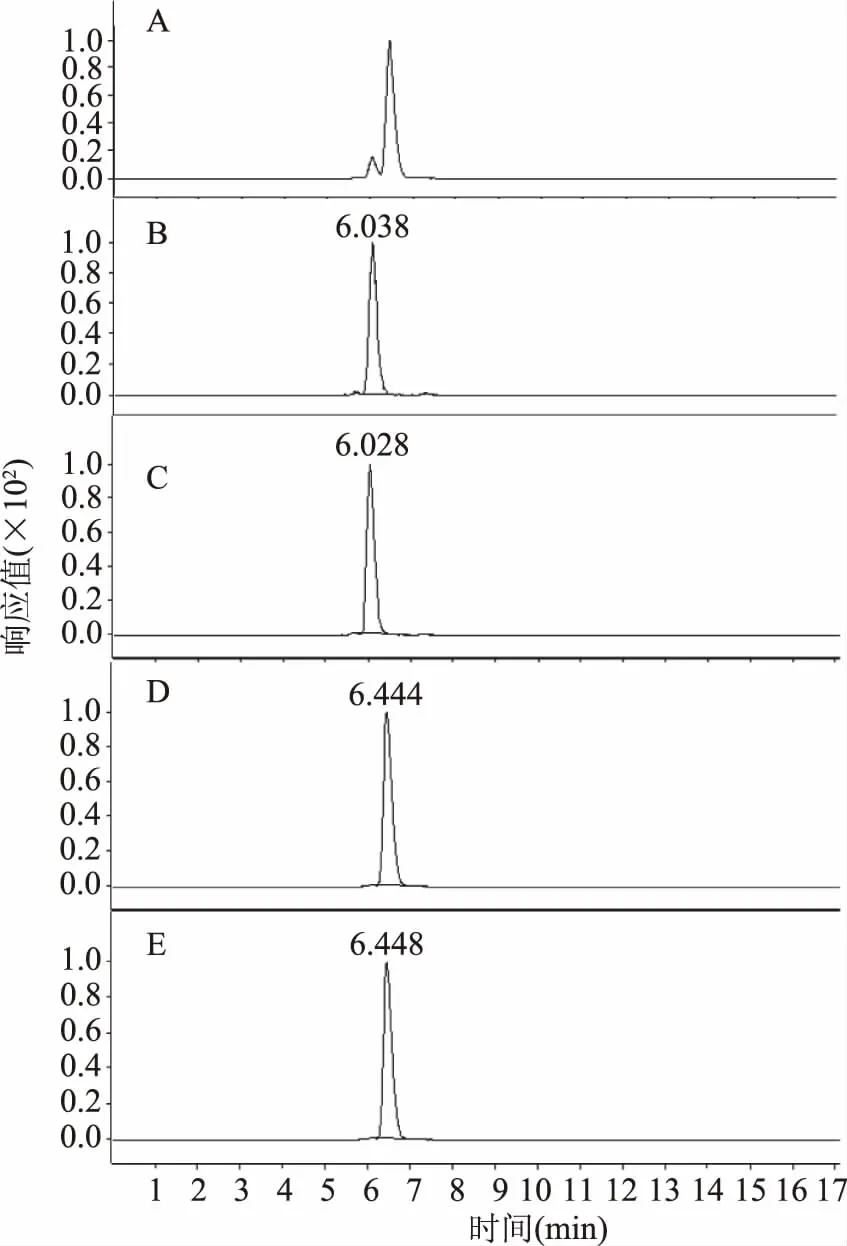
圖1 α-茄堿和α-卡茄堿標準溶液總離子及提取離子色譜圖 Fig.1 MRM chromatograms of α-solanineand α-chaconine standard solution
2.3 提取方法的優化
2.3.1 提取溶劑的優化 提取溶劑優化結果見圖2。溶劑的選擇參考國內外的相關文獻[16-27],主要是由乙酸、乙醇、水組成的體系[16-17,27],其中乙酸-水-NaHSO3(5/100/0.5,v/v/w)為美國分析化學家協會(AOAC)方法中[16]的提取溶液。
圖2 提取溶劑對馬鈴薯中茄堿提取效果的影響Fig.2 Effect of extraction solventon solanine extraction in potato
從圖2中可以看出,隨著乙酸濃度的提升,茄堿的含量也在逐步增加,這與茄堿具有弱酸性,在酸性條件下能成鹽,增加其溶解度有關。以乙酸水溶液為提取溶劑要優于乙酸乙醇;未加亞硫酸氫鈉的乙酸水溶液的提取效果更好。在酸水體系中,雖然15%乙酸水提取的茄堿含量略高,但是差異并不顯著,實驗最終選用10%的乙酸水溶液作為提取溶劑。
2.3.2 提取方式的優化 提取方式對馬鈴薯中茄堿提取效果的影響見表2。

表2 提取方式對馬鈴薯中茄堿提取效果的影響Table 2 Effect of extraction methodon solanine extraction in potato(n=3)
從表2中可以看出,勻漿提取的效果明顯高于其他的提取方式,勻漿提取3 min的提取效果大于加熱回流5 h,也高于振蕩提取2 h的效果;超聲提取40 min效果優于60 min,這可能是超聲時間過長時,超聲波的機械、空化效應可能破壞生物堿的結構[15]。實驗最終選擇勻漿3 min的提取方式。
2.3.3 料液比的優化 料液比的優化結果見圖3。從圖3中可以看出,隨著料液比的增加,樣品含量略有增加,當料液比提高至1∶10時,提取效果與料液比為1∶5時無明顯差異,考慮到不同品種馬鈴薯中茄堿含量的差異及勻漿時提取液的體積,試驗最終選擇提取料液比1∶10。

圖3 料液比對馬鈴薯中茄堿提取效果的影響Fig.3 Effect of solid-liquid ratioon solanine extraction in potato
2.3.4 提取時間的優化 不同提取時間對馬鈴薯中茄堿提取效果見圖4。從圖中可以看出,隨著提取時間的增加,α-茄堿和α-卡茄堿的含量略有增加,提取3和4 min的提取效果比較無明顯差異。考慮提取時間和提取效果等因素,實驗最終確定提取時間為3 min。
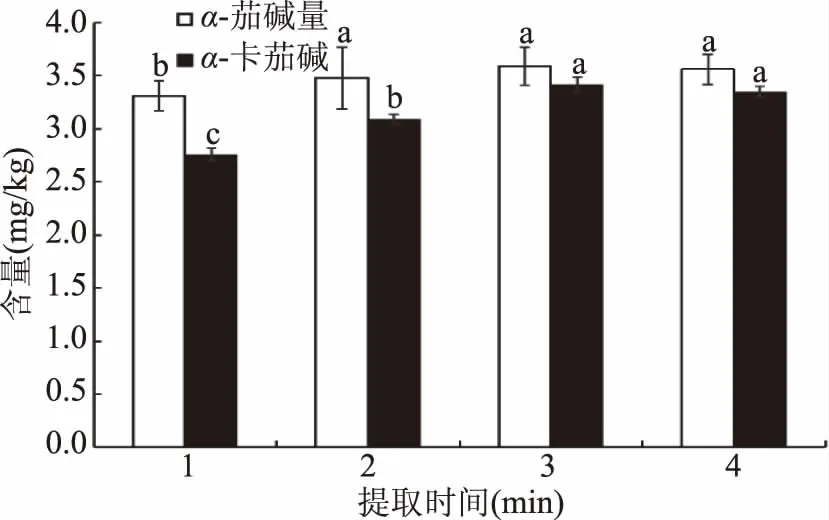
圖4 提取時間對馬鈴薯中茄堿提取效果的影響Fig.4 Effect of extraction timeon solanine extraction in potato
2.3.5 凈化條件的優化 馬鈴薯粉為加工產品,在生產過程中可能添加糖、鹽及其他化合物,為了減少基質干擾,采用C18固相萃取柱對樣品提取液進行凈化,實驗比較了不同洗脫液對洗脫效率的影響,結果見表3。
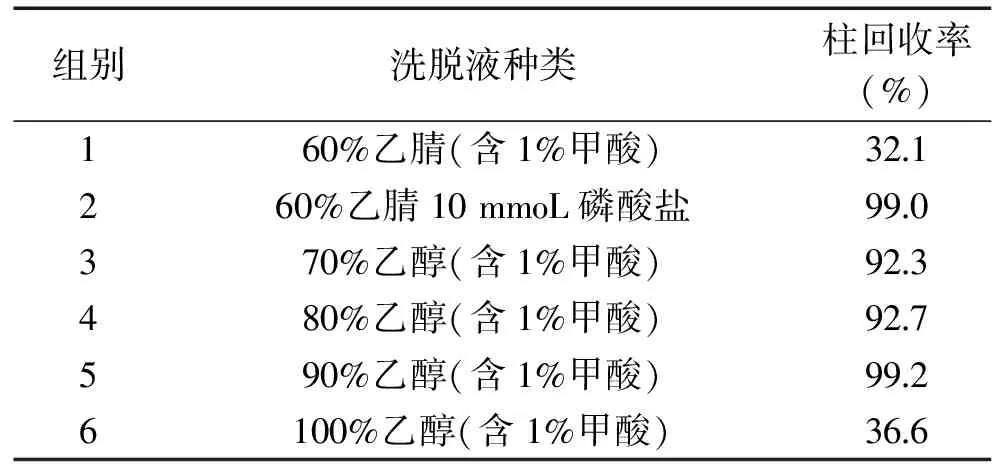
表3 洗脫液對茄堿提取效果的影響Table 3 Effect of eluent solution on solanine extraction
含磷酸鹽的洗脫液為AOAC方法中使用的,回收率較好,但該方法凈化后用高效液相色譜法檢測。為減少磷酸鹽對液相色譜質譜系統的影響,實驗將磷酸鹽緩沖液替換為水,但回收率明顯下降。將洗脫溶劑換成乙醇水體系時發現,隨著洗脫中乙醇含量的增加,柱回收有所增加,而當乙醇的含量為100%時,洗脫能力反而下降。實驗最終選擇了含90%乙醇水溶液(含1%甲酸)作為洗脫液。
2.3.6 基質效應評價 按1.2.7的濃度水平分別用溶劑和馬鈴薯及馬鈴薯粉的基質配制α-茄堿和α-卡茄堿的混合標準工作液,進行色譜-質譜分析,計算基質效應。基質效應(%)=(基質標準曲線的斜率/溶劑標準曲線的斜率-1)×100來計算。
結果表明,馬鈴薯中α-茄堿和α-卡茄堿的基質效應分別為4.73%和3.85%;馬鈴薯粉中α-茄堿和α-卡茄堿的基質效應分別為2.04%和2.15%,均小于±20%,基質效應較弱,因此可以用溶劑標準進行樣品的定量分析。
2.4 線性范圍和定量限
將混合標準工作液進入液相色譜串聯質譜中,以濃度為橫坐標,定量離子峰面積為縱坐標,繪制標準曲線。結果表明,α-茄堿和α-卡茄堿在0.01~1.00 μg/mL范圍內線性良好,α-茄堿線性方程為y=8393843x-93195,相關系數r=0.9998;α-卡茄堿線性方程為y=17650992 x-199331,相關系數r=0.9997。以10倍信噪比計算,馬鈴薯α-茄堿的定量限為0.80 μg/kg,α-卡茄堿的定量限為0.42 μg/kg;馬鈴薯粉α-茄堿的定量限為25 μg/kg,α-卡茄堿的定量限為14 μg/kg。
2.5 方法的準確度和精密度
方法的準確度采用添加回收的方式進行評價,馬鈴薯及馬鈴薯粉中α-茄堿和α-卡茄堿的添加回收率和精密度測定結果見表4。
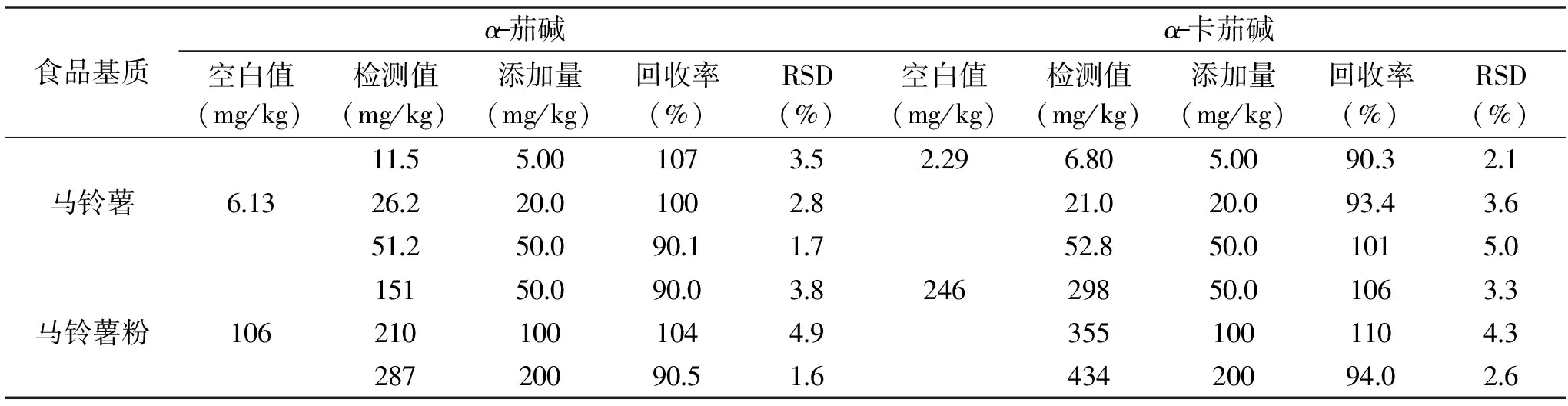
表4 馬鈴薯及馬鈴薯粉中α-茄堿和α-卡茄堿的添加率和精密度(n=6)Table 4 Recoveries and precisions of α-chaconine and α-solanine in potato and potato powder(n=6)
從表4中可以看出,馬鈴薯中α-茄堿和α-卡茄堿添加回收率在90.1%~107%之間,RSD在1.7%~5.0%之間;馬鈴薯粉α-茄堿和α-卡茄堿添加回收率在90.0%~110%之間,RSD在1.6%~4.9%之間,滿足樣品測定的要求。
2.6 市售樣品分析
采用本實驗建立的方法,對市售12個馬鈴薯樣品和3個馬鈴薯粉進行了檢測,結果見表5。從表中可以看出馬鈴薯中α-茄堿的含量在8.11~40.8 mg/kg之間,α-卡茄堿的含量在14.2~52.6 mg/kg之間。馬鈴薯粉中α-茄堿的含量在91.7~140 mg/kg之間,α-卡茄堿的含量在95.9~400 mg/kg之間。總體看α-卡茄堿的含量高于α-茄堿的含量。
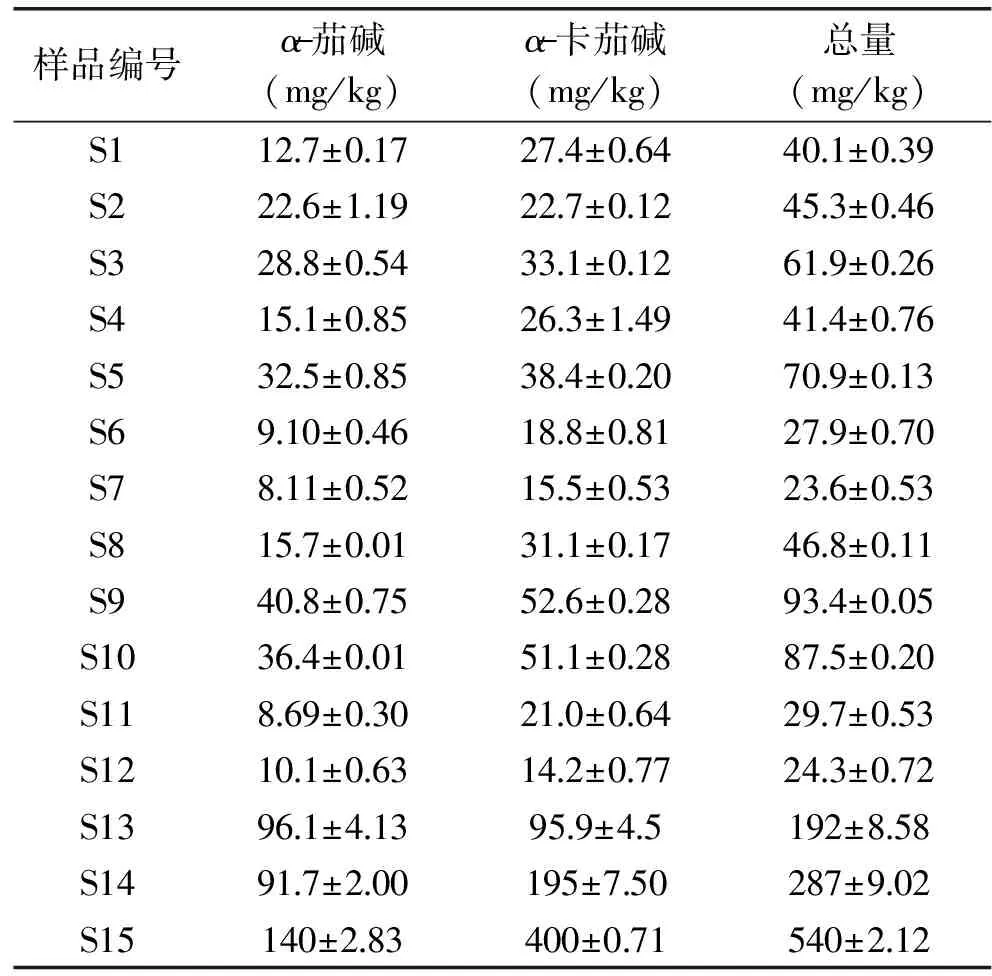
表5 市售樣品中茄堿的檢測結果Table 5 Determination of Solaninein commercial samples(n=3)
3 結論
本文建立了高效液相色譜串聯質譜法同時測定馬鈴薯及馬鈴薯粉中α-茄堿與α-卡茄堿含量的方法。方法采用勻漿提取方式,提取時間僅需3 min;在低、中、高三個添加水平的RSD均小于5%;馬鈴薯中α-茄堿和α-卡茄堿的定量限分別為0.80和0.42 μg/kg,馬鈴薯粉中α-茄堿和α-卡茄堿的定量限分別為25和14 μg/kg。方法簡單快速,重復性好,靈敏度高,可操作性強,適用于馬鈴薯及馬鈴薯粉中的α-茄堿和α-卡茄堿的同時檢測。對市售12種馬鈴薯樣品和3種馬鈴薯粉進行檢測,馬鈴薯中α-茄堿的含量在8.11~40.8 mg/kg之間,α-卡茄堿的含量在14.2~52.6 mg/kg之間;馬鈴薯粉中α-茄堿的含量在91.7~140 mg/kg之間,α-卡茄堿的含量在95.9~400 mg/kg之間。
猜你喜歡
房地產導刊(2022年5期)2022-06-01 06:20:14
能源工程(2022年1期)2022-03-29 01:06:28
建材發展導向(2021年12期)2021-07-22 08:06:48
建材發展導向(2021年7期)2021-07-16 07:07:52
中學生數理化(高中版.高二數學)(2021年12期)2021-04-26 07:43:48
中學生數理化(高中版.高考數學)(2021年12期)2021-03-08 01:28:50
今日農業(2020年16期)2020-12-14 15:04:59
消費導刊(2018年8期)2018-05-25 13:20:08
家庭影院技術(2018年4期)2018-05-09 07:07:41
電子制作(2017年20期)2017-04-26 06:57:45
