沙打旺EST-SSR分子標記開發及其遺傳多樣性分析
2019-11-18 06:18:06宮文龍王贊趙桂琴馬琳韋寶龔攀劉希強
草業學報 2019年11期
宮文龍,王贊,趙桂琴*,馬琳,韋寶,龔攀,劉希強
(1.甘肅農業大學草業學院, 草業生態系統教育部重點實驗室,中-美草地畜牧業可持續發展研究中心,甘肅 蘭州 730070;2.中國農業科學院北京畜牧獸醫研究所,北京 100193)
沙打旺(Astragalusadsurgens)原產于黃河故道地區,是一種多年生二倍體(2n=2x=16)異花授粉豆科牧草。常生長于陽坡和林緣灌叢中,主要分布在中國、前蘇聯、蒙古和北美等地[1]。因其優異的營養價值、較高的生物量和良好的適口性,現已成為我國東北、華北和西北等地區廣泛種植的牧草作物[2]。沙打旺還兼有較強的適應性、抗旱性和固沙能力等優良特性,使其在干旱、半干旱地區防止土壤侵蝕、保護自然環境和生態恢復等方面發揮越來越重要的作用[3-4]。近年來,關于沙打旺的研究主要集中在形態學、生理學和細胞學等方面[5-8]。由于受限于較長的生長周期,種質資源的遺傳改良仍為基于生長習性的表型選擇[9]。分子標記的缺乏對沙打旺種質改良和遺傳多樣性分析產生了很大限制。Huang等[10]利用19個隨機擴增多態性DNA(random amplified polymorphic DNA, RAPD)標記和96個簡單序列間重復(inter-simple sequence repeat, ISSR)標記對22個沙打旺種質的遺傳多樣性進行了綜合分析,證明了ISSR作為分析沙打旺種質遺傳多樣性的分子標記具有比RAPD更高的準確性。李瑞芬等[11]利用RAPD標記對13份不同進化狀態沙打旺種質資源的遺傳多樣性進行了調查研究,結果表明沙打旺野生材料的遺傳多樣性均高于育成品種和地方材料。然而,目前開發的有限沙打旺分子標記仍然難以滿足標記輔助育種的要求。因此,大量開發用于改良沙打旺種質的高度多態性的分子標記是十分必要的。
簡單重復序列(simple sequence repeat, SSR),也稱微衛星序列,是廣泛分布于真核生物基因組中的分子標記。按來源不同可將其分為基因組SSR和表達序列標簽SSR (EST-SSR), 由轉錄組測序得到的EST-SSR分子標記具有共顯性遺傳、多態性高、位點特異性和易檢測等優于其他分子標記的多種特點[12-13],因此廣泛應用于許多物種分子標記的開發和利用。劉歡等[14]通過聚丙烯酰胺凝膠電泳及毛細管法從200對多花黑麥草(Loliummultiflorum) EST-SSR引物中篩選出25對擴增穩定的熒光引物。剡轉轉等[15]通過白花草木樨(Melilotusalbus)轉錄組數據設計了18182對EST-SSR引物并對所開發的引物進行了篩選,為草木樨屬種質資源的遺傳改良及分子輔助育種的研究奠定了基礎。傳統的EST-SSR標記開發方法不僅效率低,而且操作復雜且成本較高,不利于大量分子標記的開發。近年來,隨著測序成本的降低,轉錄組測序成為EST-SSR分子標記開發過程中可行且重要的工具[16-18]。隨著基因編碼區中大量表達序列標簽的測定,所得到的標記不僅具有基因組SSR的特征,而且其多態性可能與基因功能直接相關[19-21]。SSR熒光標記毛細管電泳是一種基于DNA測序儀平臺的基因分型方法,與傳統的聚丙烯酰胺凝膠電泳相比,具有快速、高效、自動化、成本低、靈敏度和準確度高等優勢[22],廣泛應用于大量分子標記的開發和利用。
本研究基于轉錄組測序數據開發和驗證不同沙打旺種質中的多態性EST-SSR標記,并利用這些多態性分子標記對不同沙打旺種質的遺傳多樣性和遺傳關系進行初步研究,以期為沙打旺育種及種質改良提供良好的分析基礎和豐富的參考資源。
1 材料與方法
1.1 植物材料與DNA提取
本研究使用的27個沙打旺種質均由中國國家牧草種質庫(北京)提供(表1)。所有材料均于2017年5月種植在中國農業科學院北京畜牧獸醫研究所昌平試驗基地。試驗采用隨機區組設計,小區面積20 m2(4 m×5 m), 每小區種植一份材料,每份材料20株。植株生長6周后每份材料隨機選取5個單株幼嫩葉片組織等量混合進行DNA提取,幼葉基因組DNA采用新型植物基因組DNA提取試劑盒(天根,北京)根據說明書進行提取。通過1%瓊脂糖凝膠電泳檢測所提DNA質量,將所得模板DNA用ddH2O稀釋至50 ng·μL-1,置于-20 ℃儲存備用。
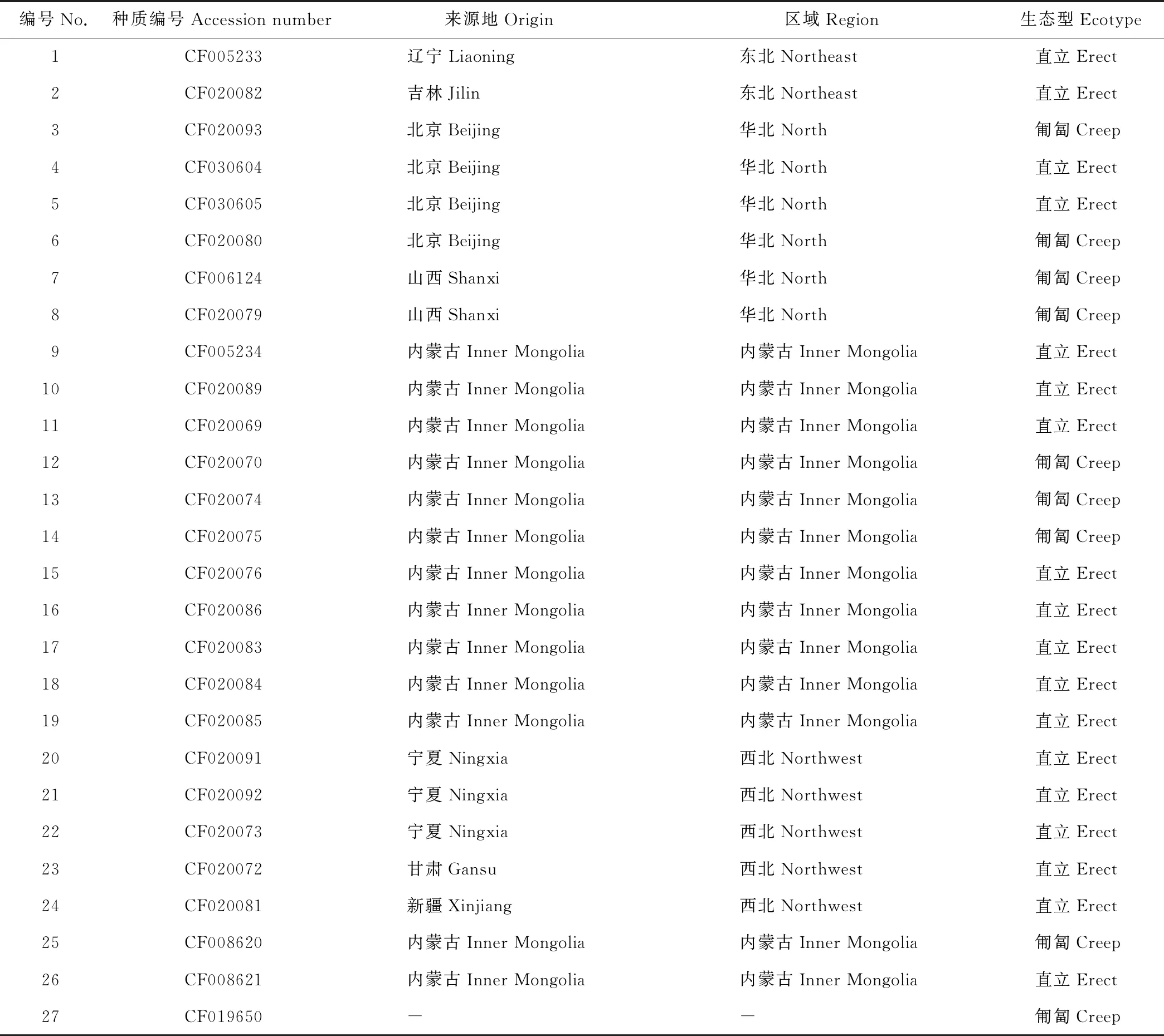
表1 供試沙打旺種質Table 1 List of all the accessions used in this study
1.2 RNA提取、cDNA文庫構建和測序、De novo轉錄組組裝和SSR位點鑒定
使用兩個沙打旺種質(CF019650, CF020070)進行RNA-seq試驗(表1)。植株生長6周后收集幼葉和莖的混合樣品(每個樣品3次重復),立即置于液氮中并儲存于-80 ℃。使用植物總RNA提取試劑盒(天根,北京)按照說明書進行樣品總RNA提取。采用多功能酶標儀(Spectra Max i3, 北京)檢測所提RNA濃度,濃度大于600 ng·μL-1的樣品用于轉錄組測序。
使用帶有Oligo(dT)的磁珠富集mRNA,在適溫下加入打斷試劑將mRNA打斷成短片段。使用隨機六聚體引物以打斷后的mRNA為模板合成第一鏈cDNA。利用緩沖液、dNTP、RNaseH和DNA聚合酶I合成第二鏈cDNA。采用Min Elute PCR純化試劑盒(天根,北京)進行純化回收、粘性末端修復和3′端加A處理。連接測序接頭,通過1%瓊脂糖凝膠電泳選擇合適大小的片段進行PCR擴增。最后,通過壹基因公司(北京)的Illumina HiseqTM2000測序平臺對構建好的cDNA文庫進行測序。再通過堿基調用圖像數據轉化為測序讀數后獲得原始reads。對reads進行過濾并去除污染序列和含有未知核苷酸比率>5%的reads從而獲得高質量的clean reads。使用默認參數的短序列裝配程序Trinity (http://sourceforge.net/projects/trinityrnaseq)進行Denovo轉錄組組裝。本研究獲得的原始序列數據已上傳至中國科學院北京基因組研究所BIG數據中心基因組序列數據庫,登記號為CRA001062。
利用簡單重復序列鑒定工具程序MISA (http://www.pgrc.ipk-gatersleben.de/misa)在unigenes數據集中對潛在的SSRs進行檢測。鑒定SSRs的標準是含有單、二、三、四、五、和六核苷酸的序列分別至少重復12、6、5、5、4和4次。
1.3 引物設計和PCR擴增
使用Batch Primer 3.0軟件進行引物設計。引物設計的主要參數為:1)引物長度為18~28 bp,最佳為23 bp;2) PCR產物大小為80~160 bp;3)退火溫度為55~65 ℃,最佳溫度為60 ℃;4) GC含量為45%~55%,最佳值為50%。本研究所用引物均由天一輝遠生物公司(北京)合成。PCR擴增采用20 μL體系:模板DNA (50 ng·μL-1) 2.0 μL, 2×Taq PCR Master Mix (10 mmol·L-1Tris-HCl, pH 8.3; 50 mmol·L-1MgCl2; 250 μmol·L-1dNTPs; 0.5 U·μL-1Taq DNA 聚合酶) 10.0 μL, 10 μmol·L-1正反引物各0.5 μL, ddH2O 7.0 μL。PCR擴增條件為94 ℃預變性5 min; 94 ℃變性30 s, 55 ℃退火45 s, 72 ℃延伸45 s; 30個循環, 72 ℃延伸10 min。通過1%瓊脂糖凝膠電泳檢測PCR產物。
隨機選擇擴增條帶清晰、具有多態性位點的引物,在每對引物5′端添加熒光標記FAM(6-carboxy-flourescein), 引物設計標準為選擇各位點引物序列間互不干擾、擴增片段長度不重疊、退火溫度相近的位點構建二重PCR。對27份沙打旺種質基因組DNA進行二重PCR擴增,熒光標記引物由華大基因公司(北京)合成。PCR反應選用25 μL體系: 10×Taq緩沖液 (100 mmol·L-1Tris-HCl, pH 8.0; 500 mmol·L-1KCl; 20 mmol·L-1MgCl2) 2.5 μL, 2.5 mmol·L-1dNTP 2.0 μL, Extaq (5 U·μL-1) 0.2 μL, 10 μmol·L-1正反引物各0.5 μL, 模板DNA (50 ng·μL-1) 1.0 μL, ddH2O 18.3 μL。PCR擴增條件為: 95 ℃預變性2 min; 95 ℃變性20 s, 54 ℃退火20 s, 72 ℃延伸30 s; 35個循環, 72 ℃延伸10 min。使用ABI3730xl DNA分析儀(美國) 進行自動熒光檢測并分離PCR產物。
1.4 數據處理
使用LIZ500分子內標(size standard)和GeneMarker v4.0軟件(http://www.appliedbiosystems.com.cn/)對毛細管電泳產物進行數據收集與圖像分析。利用PowerMarker v3.25軟件計算等位基因數,觀測雜合度(observed heterozygosity, Ho),期望雜合度(expected heterozygosity, He)和多態性信息含量(polymorphism information content, PIC)[23]。利用PowerMarker v3.25軟件計算27個沙打旺種質間遺傳距離并創建UPGMA樹形圖[23],使用MEGA 7軟件進行樹形圖繪制[24]。采用GenAlEx 6.1軟件進行主成分分析(PCoA)[25]。
2 結果與分析
2.1 EST-SSR在沙打旺轉錄組中的頻率和分布
經過嚴格的質控和數據過濾,利用短序列裝配程序Trinity共獲得151516個unigenes,總長度為190587631 bp,平均長度為1258 bp。進一步從30262個unigenes序列中檢測到39163個EST-SSR位點,SSRs分布頻率為25.85%。在含有ESTs的30262個unigenes中,6635 (21.93%)個含有兩個及以上SSR位點,3514個(11.61%)為復合型SSRs (表2)。

表2 沙打旺EST-SSR標記開發結果Table 2 Summary of EST-SSR found in erect milkvetch
從39163個EST-SSR位點的核苷酸基序長度來看:二核苷酸重復是最豐富的基序類型(38.52%),其次分別為三核苷酸(30.25%)、單核苷酸(23.04%)、五核苷酸(3.00%)、六核苷酸(2.94%)和四核苷酸(2.24%)(表3)。6個基序長度共包含1055種重復類型,其中單、二、三、四、五和六核苷酸重復分別有4、12、60、116、327和536種(表3)。

表3 沙打旺轉錄組EST-SSR基序類型及分布特征Table 3 Distribution of the EST-SSR motifs in transcriptome
不同EST-SSR基序長度中的重復頻率和主要重復基序通常是不同的。本研究中單、二、三、四、五和六核苷酸基序的主要重復頻率分別為12~15、6~11、5~8、5~6、4~6和4~6次(圖1)。單核苷酸基序類型中最豐富的重復基序是A/T,其包含95.37%的單核苷酸重復類型,其次為AG/CT (包含71.39%的二核苷酸重復),AAG/CTT (包含29.18%的三核苷酸重復),AAAT/ATTT (包含28.73%的四核苷酸重復),AAGAG/CTCTT (包含9.19%的五核苷酸重復)和AAGATG/ATCTTC (包含3.73%的六核苷酸重復)(表3)。
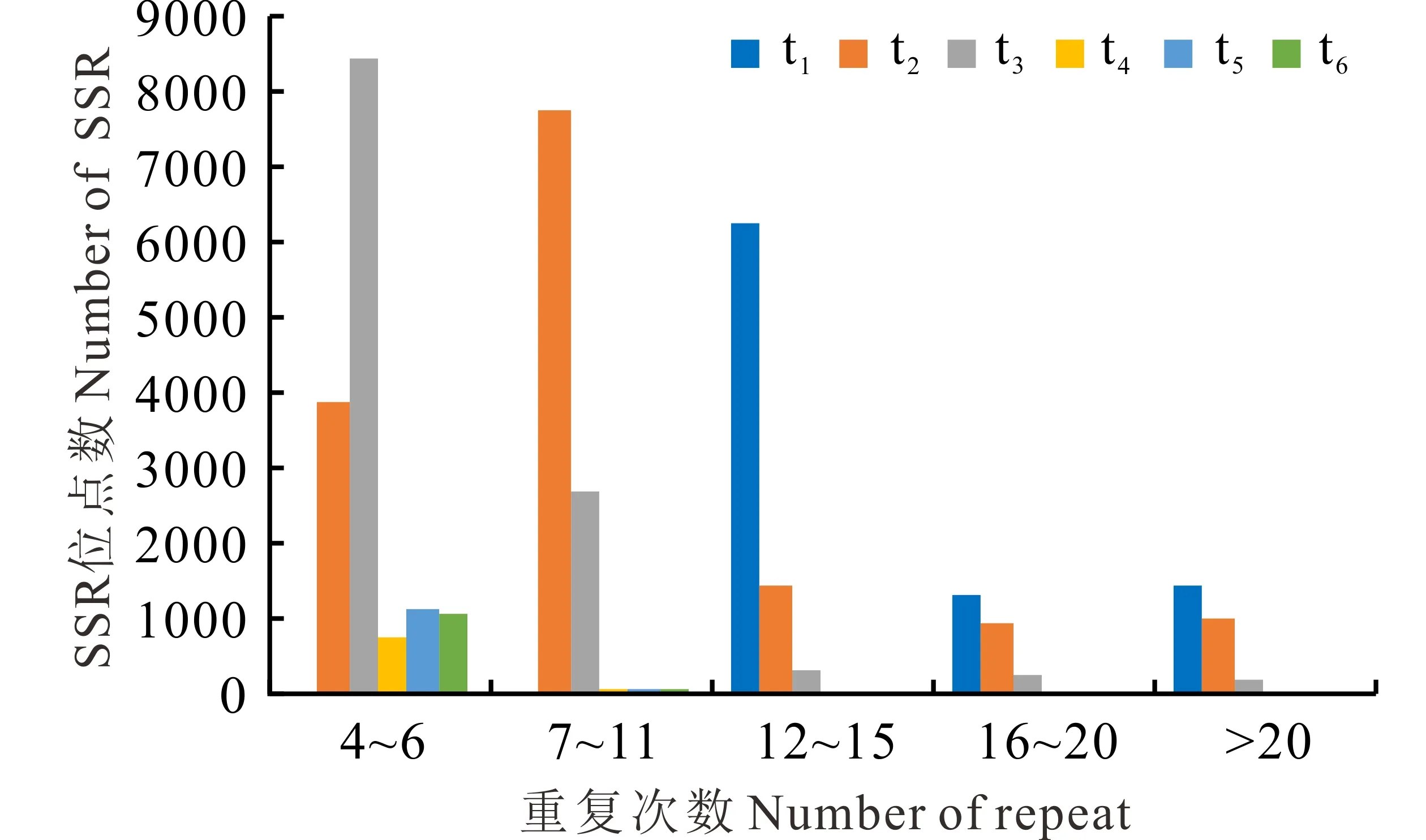
圖1 SSR重復單元及重復次數的分布Fig.1 The distribution of SSR motifs and repeat number
所有沙打旺轉錄組SSRs序列的平均長度為26.16 bp,單、二、三、四、五和六核苷酸重復的平均長度分別為19.07、27.13、25.25、27.60、26.20 和31.68 bp。其中分布在12~15 bp的最多,占總數的42.92%,其次為16~19 bp (16.60%)、≥32 bp (16.35%)、20~23 bp (13.55%)、24~27 bp (8.09%)和28~31 bp (2.49%)(圖2)。
2.2 EST-SSR多態性引物篩選及遺傳多樣性分析
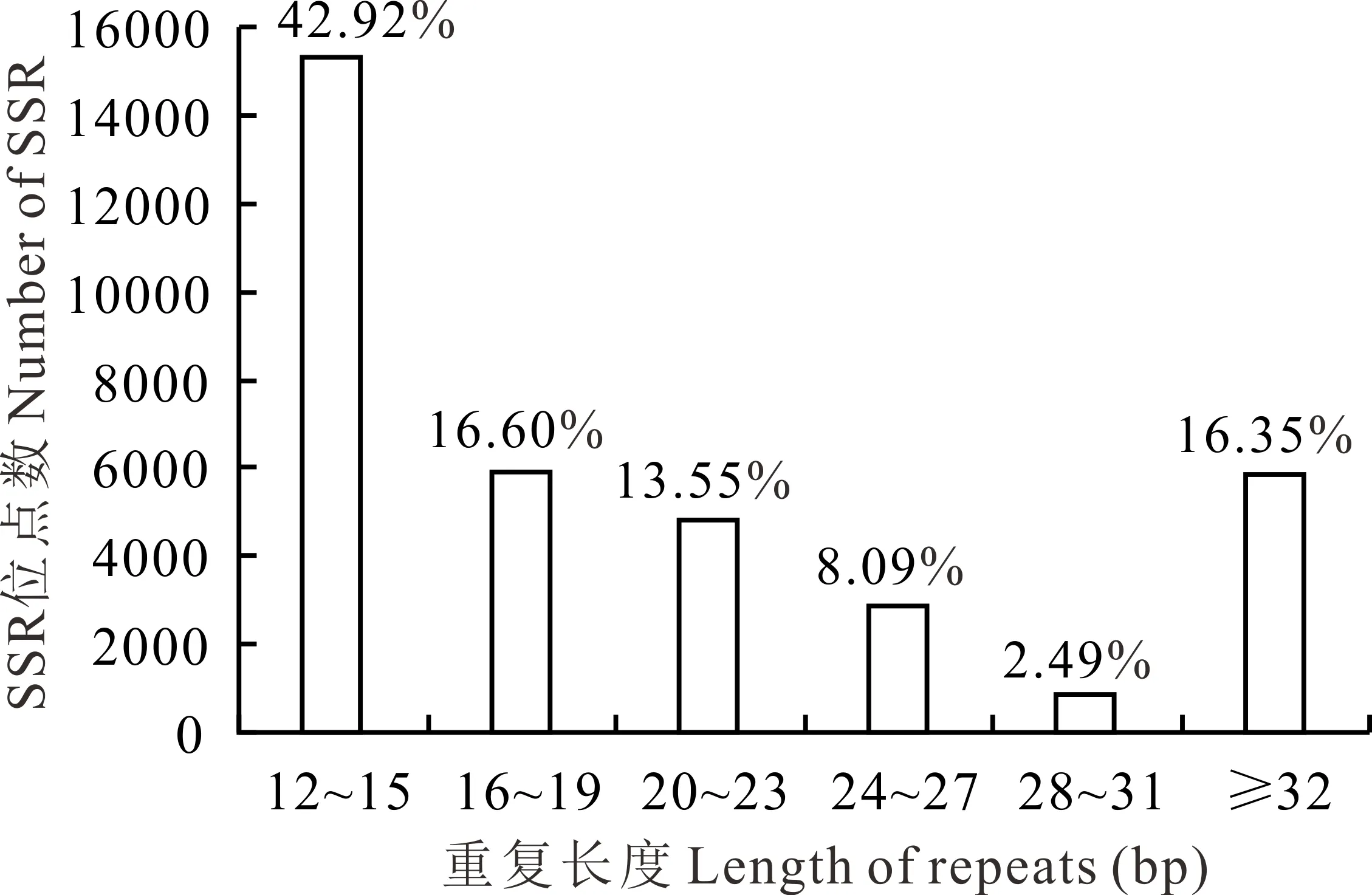
圖2 SSR重復長度的分布Fig.2 Distribution of the SSR repeat length
本研究從30262個unigenes中成功設計了22367對引物,剩余的ESTs由于SSR的側翼序列太短(<40個核苷酸)或不符合引物設計的標準從而未能成功進行引物設計。用兩個沙打旺種質(CF019650,CF020070)的基因組DNA對隨機選擇的100對引物(除單核苷酸重復)進行初步篩選,其中90對引物(90%)可擴增出目的特異性條帶,剩余10對引物產生的擴增條帶均不符合預期大小。進一步從成功擴增的90對引物中隨機選擇51對對27個沙打旺種質基因組DNA進行基因分型(表4和圖3)。
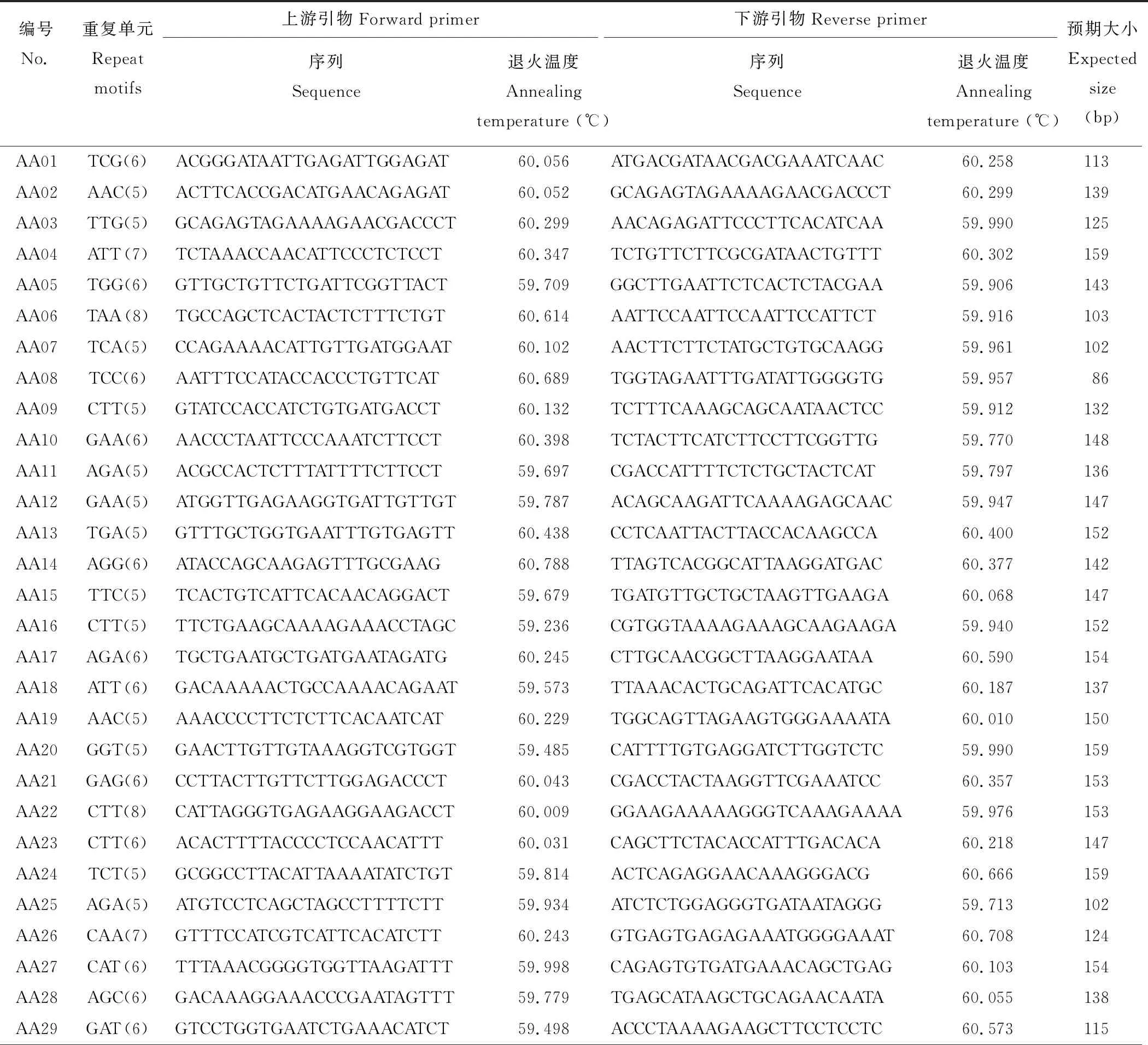
表4 用于基因分型的引物序列及信息Table 4 Primer sequences and information for genotyping in this study
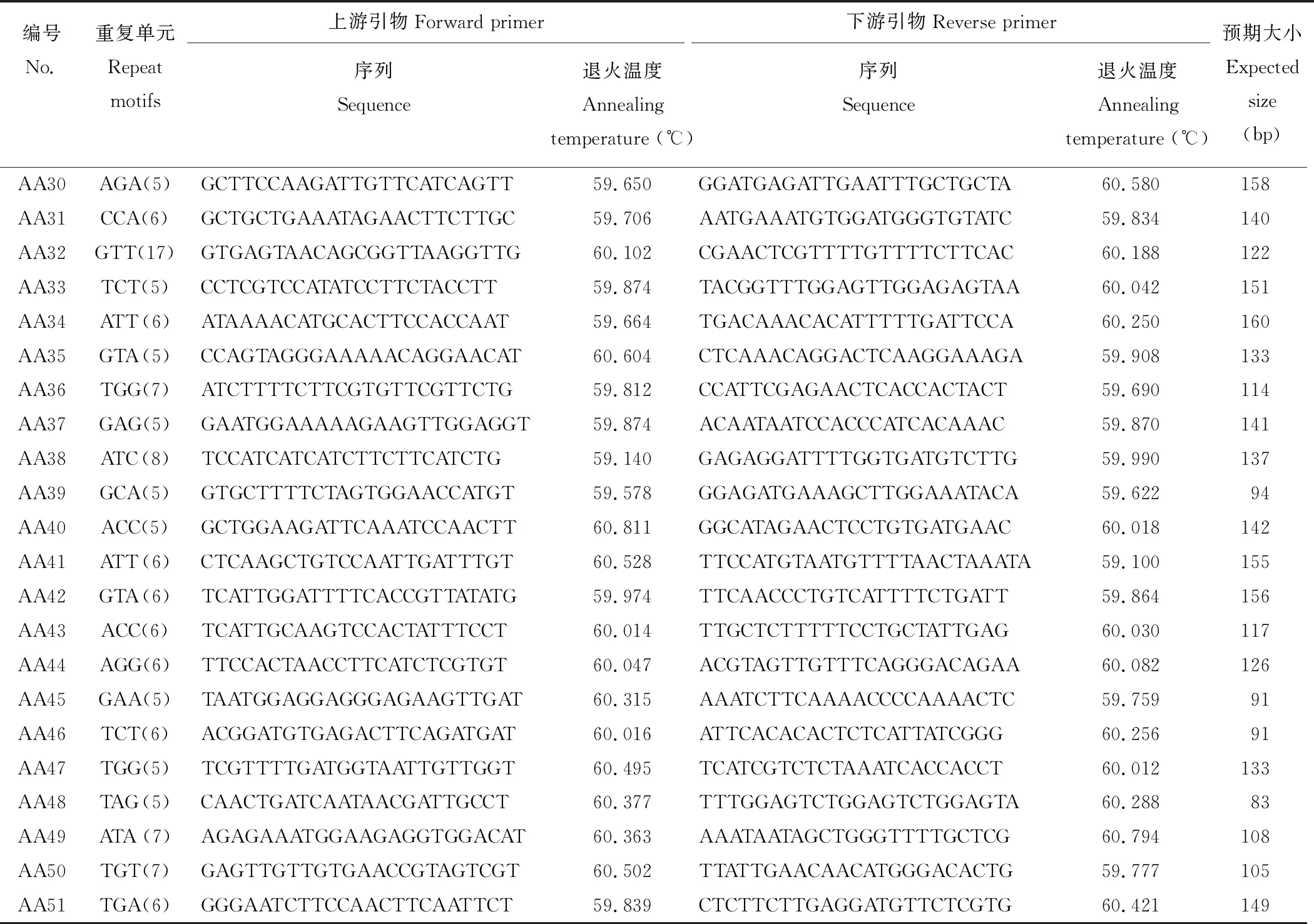
續表4 Continued Table 4
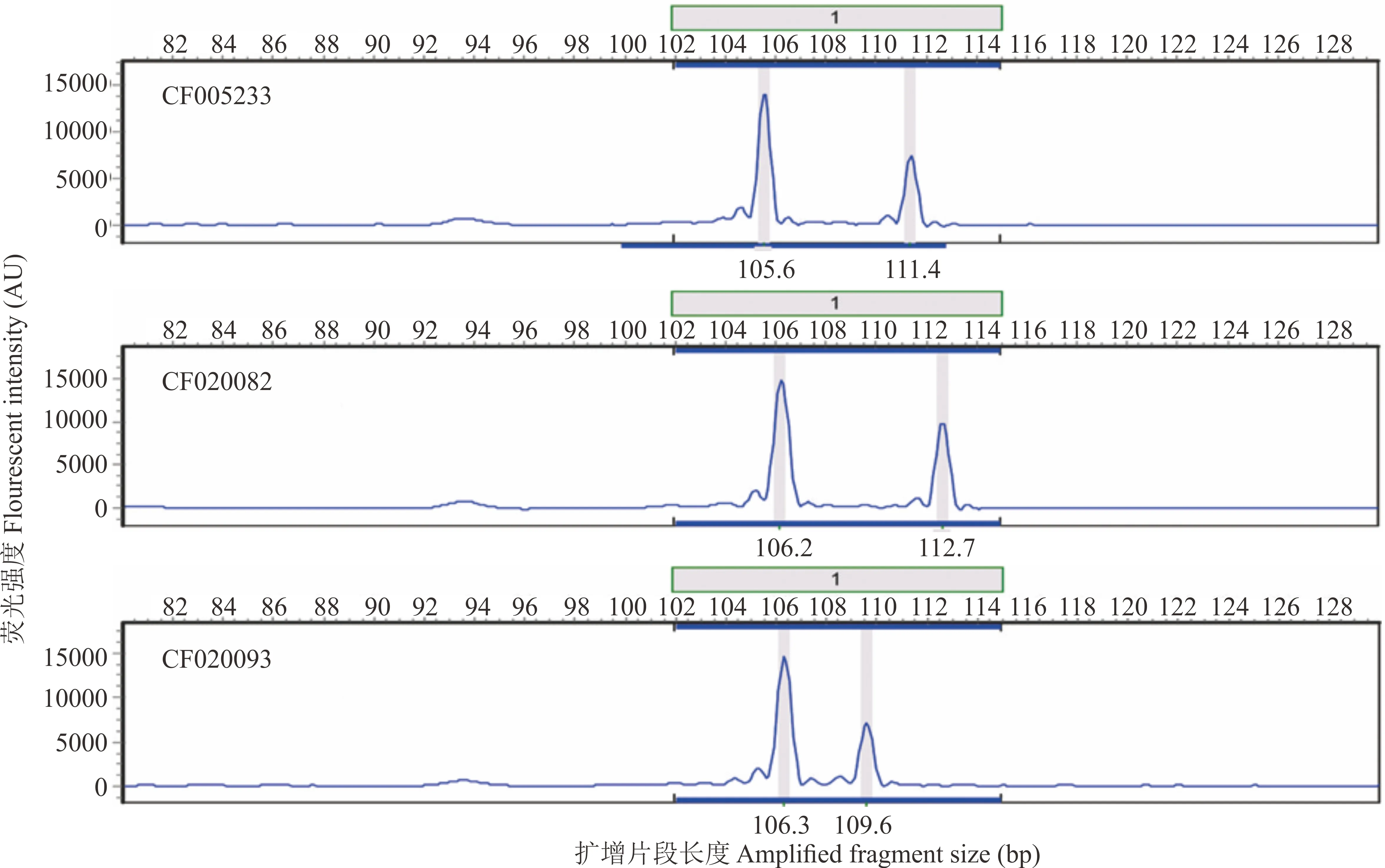
圖3 3個沙打旺種質在AA01位點的電泳圖譜分析Fig.3 Electrophoretogram analysis of one EST-SSR loci (AA01) among 3 erect milkvetch accessions
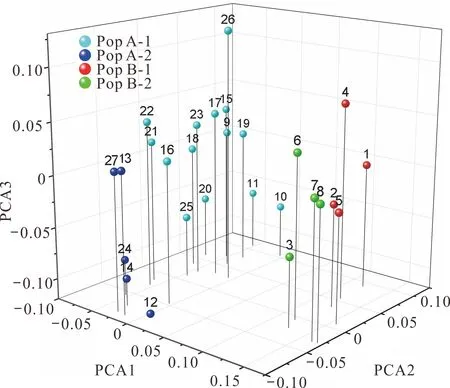
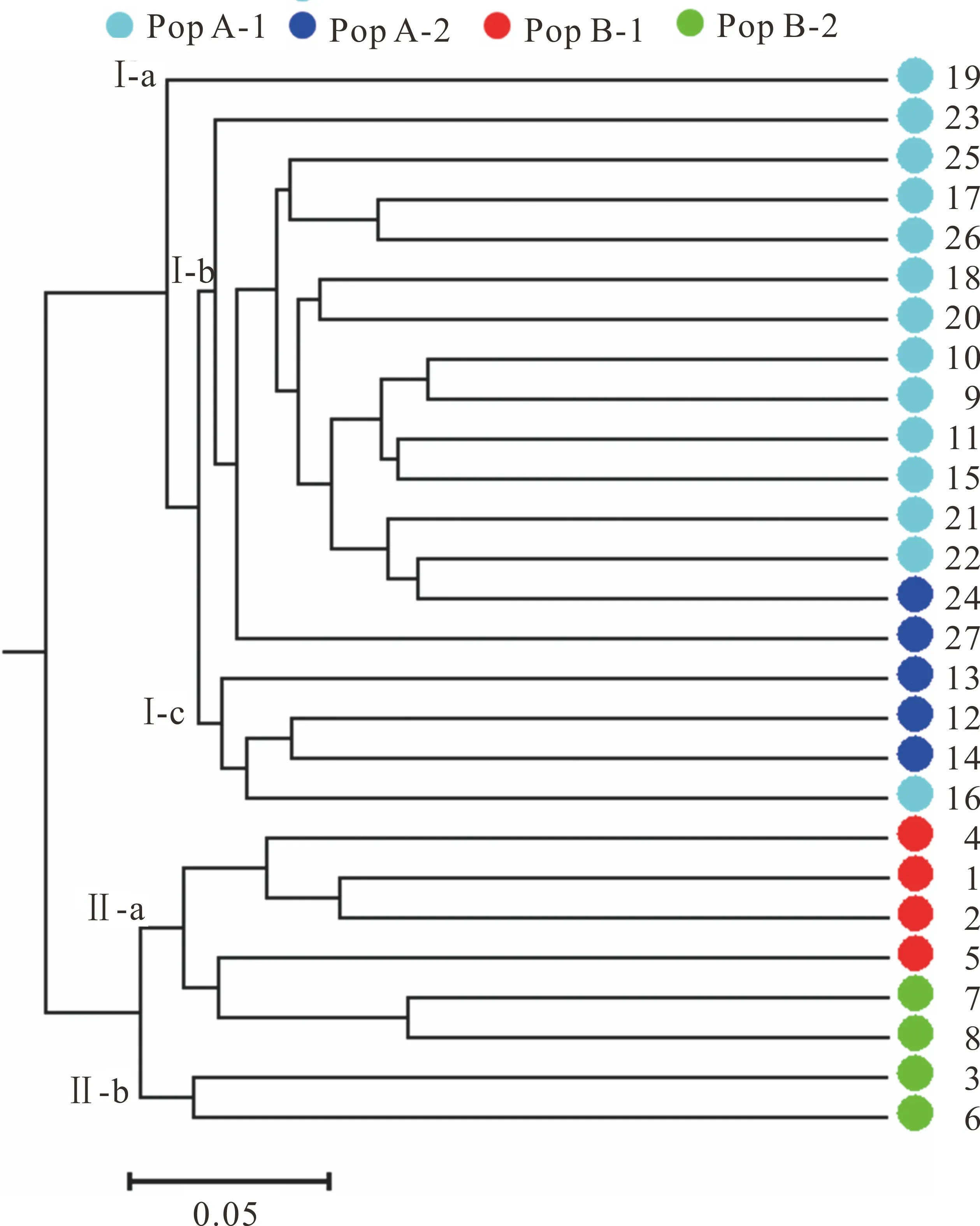
基因分型結果顯示:51個位點共檢測到446個等位基因,每個EST-SSR位點平均檢測到8.75個等位基因,變化范圍從2個(AA26)到17個(AA19)。期望雜合度(He)變化范圍為0.235 (AA10)~0.906 (AA38),平均值為0.719。觀測雜合度(Ho)為0.259 (AA10)~1.000 (AA06),平均值為0.730。多態性信息含量(PIC)為0.224 (AA10)~0.898 (AA38),平均值為0.682 (表5)。根據Botstein等[26]對多態性的定義,51個標記中有45個具有高度多態性(PIC>0.50),5個具有中度多態性(0.25 計算27個沙打旺種質間的遺傳距離并進行主成分分析(PCoA)(圖4),結果顯示前3個主成分分別解釋了變異的13.93%,7.09%和6.35%,占總變異的27.37%。第一主成分(PCo1)可根據地理分布將27個種質分為兩個類群(Pop A和Pop B)。Pop A包括19個種質(13個來自內蒙古,5個來自西北,1個來自未知地區),而Pop B共包括8個種質(6個來自華北,2個來自東北)。通過第二主成分(PCo2)可將Pop A和Pop B進一步劃分為4個子群(Pop A-1, Pop A-2, Pop B-1, Pop B-2)。由圖4可知,這4個子群分別主要由具有直立或匍匐生態型的種質組成:Pop A-1包括14個種質(13個直立種質和1個匍匐種質),Pop A-2包括5個種質(1個直立種質和4個匍匐種質),Pop B-1和Pop B-2分別包括4個直立和4個匍匐種質(圖4)。表明地理分布和生態型可以在很大程度上反映沙打旺種質的遺傳特征差異。 圖4 27個沙打旺種質主成分分析Fig.4 Three-dimensional principal coordinate analysis (PCoA) of 27 erect milkvetch accessions 圖5 27個沙打旺種質UPGMA 聚類分析Fig.5 UPGMA dendrogram of 27 erect milkvetch accessions 為進一步評估沙打旺種質之間的遺傳關系,利用Nei遺傳距離構建了27個種質的UPGMA樹形圖(圖5)。UPGMA聚類結果與PCoA分析基本一致。首先確定了兩大類群Ⅰ和 Ⅱ,結合圖4和圖5可知,Pop A和Pop B中的種質分別位于聚類組Ⅰ和聚類組Ⅱ中。聚類組Ⅰ可以進一步分為3個亞組,即Ⅰ-a,Ⅰ-b和Ⅰ-c。Ⅰ-a僅包含1個種質(CF020085)。而來自Pop A-1的大多數種質位于Ⅰ-b (86%)中,其余種質平均分布在Ⅰ-a (7%)和Ⅰ-c (7%)。Pop A-2的大部分種質都位于Ⅰ-c中(60%),40%位于Ⅰ-b中 (圖5)。聚類組Ⅱ也可以進一步分為兩個亞組,即Ⅱ-a和 Ⅱ-b。來自Pop B-1的種質全部位于Ⅱ-a中(100%),而來自Pop B-2的種質則分別位于Ⅱ-a (50%)和Ⅱ-b (50%)中(圖5)。聚類結果表明雖然在少數情況下,來自4個子群的部分種質沒有在5個亞組中聚集在一起,但在大多數情況下大部分種質的聚類結果仍然與其生態型及地理來源具有較高相關性。 Denovo轉錄組測序已被證明是一種有效且準確的EST-SSR標記開發和鑒定方法,并已成功應用于許多植物物種中[27-28]。本研究利用Denovo轉錄組測序從沙打旺的30262條unigenes序列中成功鑒定出39163個EST-SSR位點,SSRs分布頻率為25.85%,高于之前在苜蓿(Medicagosativa)[29]和草木樨(Melilotus)[30]中的報道。SSRs重復基序的類型在不同物種之間通常是不同的,但在大多數物種中最豐富的重復基序(除單核苷酸重復外)均為二核苷酸和三核苷酸,這可能與物種的進化歷史和基序的基因表達程度有關[31-32]。本研究中,最豐富的基序類型同樣是二核苷酸重復,占總重復的38.52%,這與白菜(Brassicacampestris)[33]、珙桐(Davidiainvolucrata)[34]和橡膠樹(Heveabrasiliensis)[35]的研究結果一致。其次最常見的基序類型是三核苷酸重復,占總重復的30.25%,也是黃羽扇豆(Lupinusluteus)[36]、老芒麥(Elymussibiricus)[37]和刺槐(Robiniapseudoacacia)[19]中最豐富的重復基序。此外,AG/CT和AAG/CTT分別是二核苷酸和三核苷酸重復中最常見的重復單元,這與橡膠樹[35]和檸條錦雞兒(Caraganakorshinskii)[38]的研究結果相同。 針對所有EST-SSR位點進行引物設計,成功獲得了22367對特異性引物,這為沙打旺遺傳育種研究提供了豐富的資源。利用兩個沙打旺種質基因組DNA對隨機選擇的100對引物進行初步篩選,其中90對引物可成功擴增出目的特異性條帶,這一結果高于寧夏枸杞(Lyciumbarbarum)[18]和苜蓿[29]中的成功擴增比例。較高的擴增率可能是因為本研究中使用的種質均屬于具有相似遺傳結構的沙打旺種而不涉及其他近緣物種。利用篩選到的多態性引物分析27個沙打旺種質的遺傳多樣性,我們在51個EST-SSR位點共檢測到446個等位基因,平均He、Ho和PIC分別為0.719、0.730和0.682,均高于之前在苜蓿[39]、牛角屬(Calotropis)[40]和蔥屬(Allium)[41]中的報道。上述結果表明本研究中開發的EST-SSR標記具有較高水平的多態性,適用于沙打旺遺傳和育種的研究和應用。此外,傳統上使用聚丙烯酰胺凝膠電泳進行基因分型已被證明是低效且不準確的[42],本研究中的51個EST-SSR位點均通過自動DNA分析平臺進行基因分型,檢測結果更加靈敏準確。 遺傳關系分析可以揭示特定種質的遺傳多樣性,并可用于標記輔助育種[10]。以前的許多研究都集中在對部分地區沙打旺種質之間的遺傳關系進行研究和分析[10-11],而本研究包括了來自中國各地區的種質,結果更加具有代表性和準確性。通過UPGMA聚類分析將沙打旺種質分成兩個類群,其中一個類群包括來自西北和內蒙古的種質,另一個類群主要包括來自華北和東北的種質,這可能是由于臨近的地理區域和長期的人工選擇導致在兩個群體中形成了相似的遺傳關系。PCoA分析基于直立和匍匐兩種生態型將大部分種質(除CF020081和CF008620)劃分為4個子群,這意味著兩種不同的生態型可以在很大程度上反映不同沙打旺種質的遺傳特征。其余兩個種質(CF020081, CF008620)未根據生態型聚集在一起,可能是因為長期的自然選擇導致其遺傳背景發生了部分改變。UPGMA聚類結果與PCoA分析結果基本一致,然而并非所有的種質都基于其地理來源在5個亞組中聚集在一起,這可能與鄰近地區長期的人工選擇和種質交換有關。為更全面的了解不同沙打旺種質間的遺傳關系,有必要使用來自中國乃至世界各地的大量不同種質進行遺傳多樣性及遺傳關系評估,以便為沙打旺種質的改良和利用提供行之有效的手段和重要的參考資源。 本研究通過Denovo轉錄組測序從30262個unigenes中鑒定了39163個EST-SSR位點,成功設計了22367對引物,并對100對引物進行驗證共篩選出51對多態性引物,這為沙打旺乃至黃芪屬(Astragalus)物種的分子標記開發及利用提供了良好的基礎。此外,對27個沙打旺種質的遺傳多樣性進行了初步分析,結果表明本研究開發的多態性EST-SSR分子標記具有較高水平的遺傳多樣性,有助于沙打旺分子標記輔助育種,QTL定位和遺傳變異研究。主成分和聚類分析表明不同沙打旺種質之間的遺傳關系與其地理來源具有較高的相關性,且不同生態型(直立或匍匐)種質的遺傳分布具有明顯的種質特異性,為沙打旺種質改良和遺傳多樣性研究提供了重要的參考資源。2.3 主成分和聚類分析
3 討論
4 結論
