分散固相萃取結合氣相色譜- 四極桿- 飛行時間質譜測定茶葉中13種三唑類農藥殘留
2019-11-04 08:41:58滕曉宇李建勛胡雪艷龐國芳范春林
食品科學技術學報 2019年5期
關鍵詞:檢測
滕曉宇, 李建勛, 胡雪艷, 龐國芳, 范春林,*
(1.北京工商大學 食品與健康學院, 北京 100048; 2.中國檢驗檢疫科學研究院, 北京 100176)
三唑類農藥應用廣泛,大多數屬于中低毒性農藥,但其廣譜程度高、持效期長、內吸性強[1],有引起慢性中毒的風險[2]。
茶葉的生產和使用在我國具有悠久的歷史,其食用方式多為直接沖泡,缺少一般食物烹飪的持續高溫等步驟,這樣也會造成茶葉中農藥殘留無法有效揮發或降解失活[3]。歐盟和日本針對農藥殘留的限量標準最為嚴苛[4],文章所涉及的13種三唑類農藥在歐盟MRL(最高殘留量)標準中均有其在茶葉中的嚴格規定。建立準確、靈敏的檢測方法不僅可以為國民身體健康提供技術保障,也有利于突破國際貿易壁壘。在農藥殘留的前處理技術中,主要有液液萃取、固相萃取和加速溶劑萃取等方法[4-5],其中固相萃取由于具有優異的去除雜質的效果和良好的重復性得到了廣泛的應用,如我國現行標準GB/T 23204—2008[5]中使用了Cleanert TPT固相萃取柱進行凈化。該方法具有凈化效果好、準確度高的優點,但有機溶劑用量較大。因此,文章旨在建立一種達到歐盟MRL標準檢測要求的茶葉中13種三唑類農藥的分散固相前處理和檢測方法[6-9],為將來開發更靈敏、環保的農藥殘留檢測技術提供實驗依據。
1 材料與方法
1.1 材料與試劑
茶葉樣品,宏德遠利市場。糠菌唑、環丙唑醇、烯唑醇、氟硅唑、粉唑醇、己唑醇、葉菌唑、多效唑、戊菌唑、戊唑醇、四氟醚唑、三唑酮、三環唑標準品,純度均大于95%,德國Dr. Ehrenstorfer公司;N-丙基乙二胺(PSA)吸附劑(40~60 μm)、石墨化炭黑(GCB)吸附劑(120~400 mesh)、多壁碳納米(Cleaner NANO-carb)(10~20 nm),天津Bonna-Agela公司;乙腈、正己烷,色譜純,美國Thermo公司;醋酸,分析純,北京化學試劑公司;尼龍66濾膜,天津津騰實驗設備有限公司。
1.2 儀器與設備
7890A- 7200型氣相色譜- 四極桿- 飛行時間質譜儀,美國安捷倫科技公司;SR- 2DS型水平振蕩器,日本Taitec公司;X- 30R型落地式冷凍離心機,貝克曼庫爾特商貿(中國)有限公司;N- EVAP112型氮吹儀,美國OA- SYS公司;R- 215型旋轉蒸發儀,瑞士Buchi公司;TM- 1FN型渦旋混勻器,日本Asone公司。
1.3 實驗方法
1.3.1農藥標準溶液的配制
準確稱取10 mg(精確至0.01 mg)農藥標準品分別于10 mL容量瓶,用甲醇溶解定容至刻度配制成質量濃度為1000 mg/L的標準工作溶液,靜置過夜后于25 mL容量瓶配制成質量濃度為10 mg/L的混合標準工作溶液。
1.3.2樣品前處理
茶葉樣品粉碎后過16目篩取得粉末。稱取5.0 g粉末于50 mL帶蓋聚丙烯離心管中,加入10.0 mL體積分數1%醋酸乙腈均質提取1 min,以1×104r/min離心5 min,取5 mL上清液于另一個裝有45 mg NANO- carb、75 mg PSA、25 mg GCB吸附劑的15 mL離心管中,以約2 200 r/min渦旋1 min混勻,以1×104r/min離心3 min。移取4 mL上清液至80 mL雞心瓶中,在40 ℃旋蒸至約0.5 mL,氮吹至近干,0.5 mL正己烷定容,過膜用于氣相色譜- 四極桿- 飛行時間質譜儀分析。
1.3.3色譜條件
色譜柱:VF- 1701 ms毛細管柱(30 m×0.25 mm×0.25 μm);程序升溫條件:初始柱溫為80 ℃,以40 ℃/min升溫至220 ℃,再以8 ℃/min升溫至280 ℃,保持2 min;載氣:氦氣,純度≥99.999%,流速1.2 mL/min;進樣口溫度:270 ℃;傳輸線溫度:280 ℃;進樣量:1 μL;進樣方式:不分流進樣。
1.3.4質譜條件
電子轟擊(EI)離子源。電子能量為70 eV,燈絲電流為50 μA,離子源溫度為230 ℃,掃描模式為全掃描,質量掃描范圍(m/z)為50~600 u,采集速率為2 spec/s,溶劑延遲時間為6 min。
1.3.5精確質量數數據庫的建立
在全掃描模式下采集,建立13種農藥的個人化合物數據庫及譜庫(personal compound database and library,PCDL),定性、定量離子具體信息見表1。
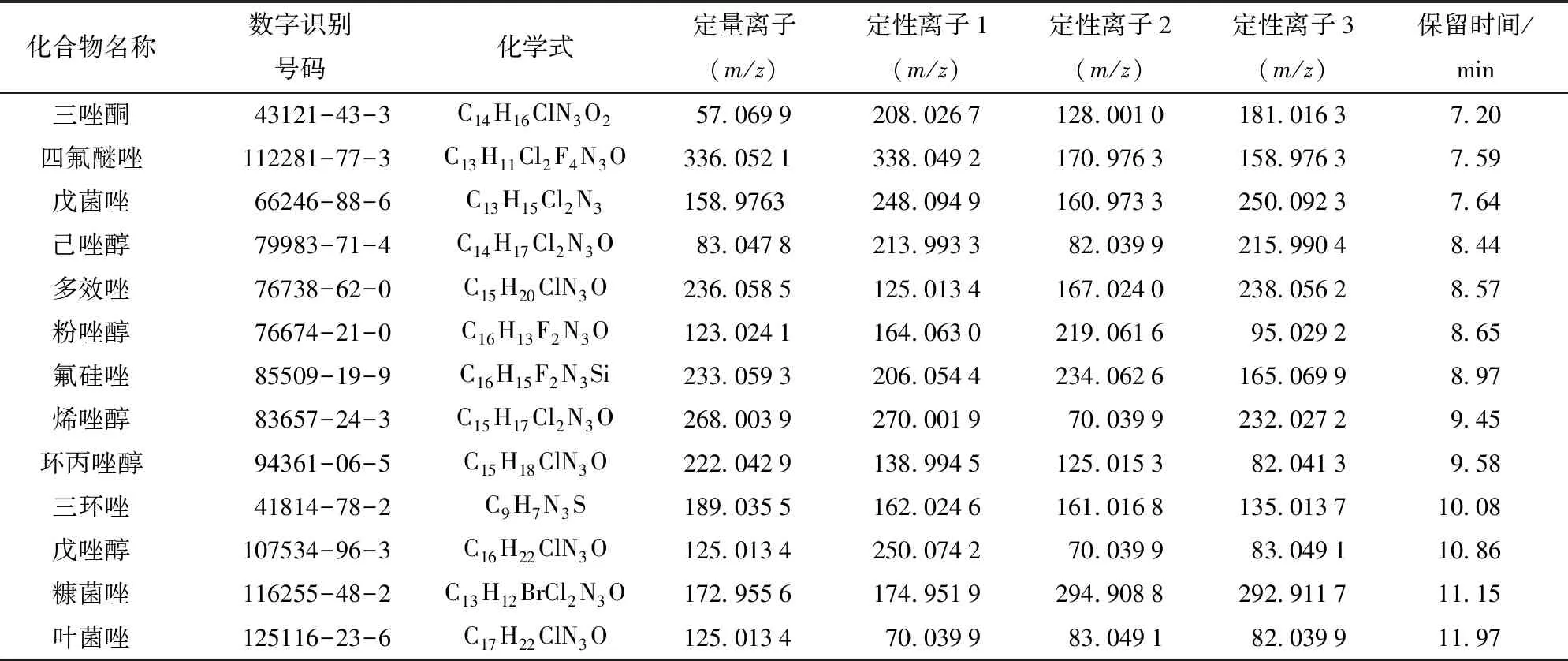
表1 13種農藥的名稱、CAS號、化學式及質譜參數Tab.1 Compound names, CAS numbers, formula and mass references of 13 pesticides
1.4 數據處理
使用儀器自帶軟件Mass Hunter B.07進行數據處理。
2 結果與分析
2.1 色譜質譜條件優化
為了提高分離效率,縮短檢測時間,對氣相色譜升溫程序進行優化,使13種農藥的出峰時間介于7.1~12.2 min。
13種農藥的總離子流色譜見圖1,與PCDL庫對比定性,按照序號從小到大依次為三唑酮、四氟醚唑、戊菌唑、己唑醇、多效唑、粉唑醇、氟硅唑、烯唑醇、環丙唑醇、三環唑、戊唑醇、糠菌唑、葉菌唑。其中12號的糠菌唑經質譜圖鏡像對比確證為雙峰。
2.2 提取方式優化
參考綜述文獻中所用的提取方法[3],采取渦旋、振蕩、均質、超聲4種方式進行提取。但在本實驗中渦旋方式無法使茶葉中殘留農藥獲得有效的回收率,這可能是茶葉粉末質地緊密、細致,提取能量不足以使提取液充分滲透溶解基質中的農藥殘留導致的,故棄去渦旋提取的方法,其余提取方法具體為:1)均質提取,以1.4×104r/min提取1 min,每個樣品均質后分別用水和乙腈3次清洗刀頭。2)超聲提取,室溫下樣品置于超聲清洗機中水浴,分別提取1、3、5 min。3)震蕩提取,室溫下樣品置于水平震蕩機,震蕩速度150 r/min,分別提取5、10、30 min。
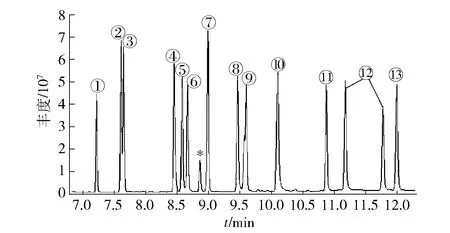
*表示干擾離子。圖1 13種農藥的總離子流色譜Fig.1 Total ion chromatogram of 13 pesticides
所有提取方式的回收率均在71%~102%,農藥回收率的相對標準偏差(RSD)除震蕩30 min提取的三唑酮以外,其余提取方式均小于20%(見表2)。
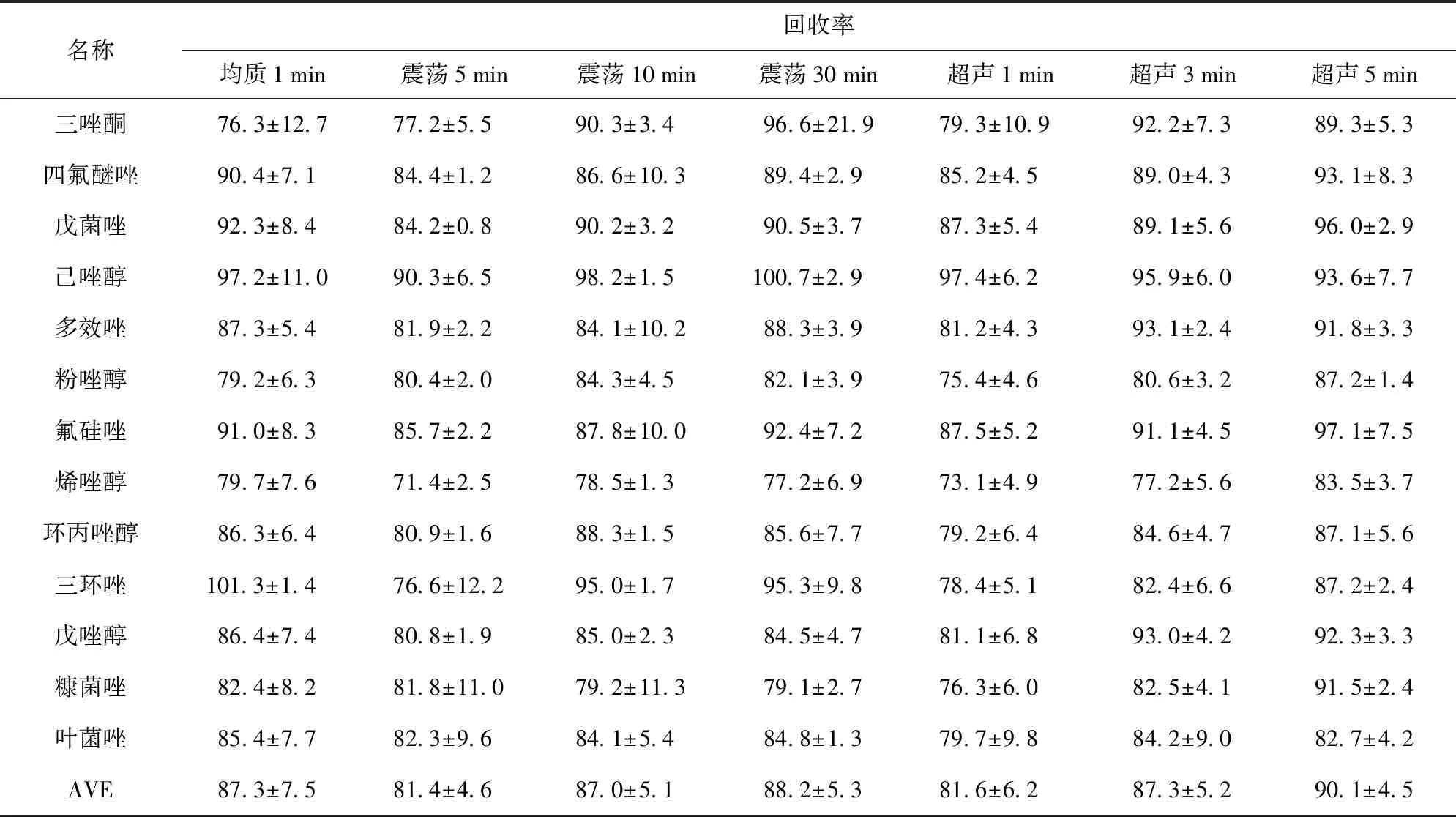
表2 不同提取條件下13種農藥的回收率Tab.2 Recoveries of 13 pesticides under different extract conditions %
n=5。
超聲5 min提取效果最好,平均回收率為90.1%;均質1 min,震蕩10、30 min、超聲3 min的提取效果比較接近,效果也比較好,平均回收率在87%~88%左右;其余2種略差為81%左右。綜合考量提取效果、時間、操作簡便程度等方面,最終選取均質1 min作為前處理提取方法。
2.3 凈化填料的配比選擇
Hayward等[10]進行了含戊唑醇在內的227種農藥在紅、綠、白、烏龍茶中的添加回收驗證,使用加入不同比例的PSA與GCB的ECPSA固相萃取柱和TPT固相萃取柱以及加入了PSA與carbon X Plus的COA固相萃取柱進行凈化,經氣相色譜- 串聯質譜檢測,對比統計結果顯示:3種固相萃取柱中,含有500 mg PSA和500 mg GCB的TPT固相萃取柱較其余2種表現出更好的凈化回收效果。Hou等[11]在分散固相萃取的過程中,使用NANO- carb和PSA等凈化材料,建立了茶葉中含三唑酮在內的78種農藥殘留的氣相色譜- 三重四極桿聯用質譜分析方法,研究中通過對NANO- carb和PSA的配比優化,提高了共提物的凈化作用,獲得了良好的線性范圍和檢出限。王學翠等[12]利用高效液相色譜法對牛奶中6種四環素進行檢測,結果顯示:多壁碳納米管基質固相分散萃取牛奶中四環素的回收率優于C18,可推廣應用到食品等復雜樣品中其他農獸藥的殘留分析。
本實驗的基礎凈化填料使用了常用來去除有機酸、金屬離子、酚類、蠟質和色素的PSA和GCB[13-14]填料,還使用了近十年來使用頻率越來越高的新型凈化材料—多壁碳納米管[15-17],這種納米材料的多層平行網狀結構,具有優質的吸附性和穩定性[18]。參考上述文獻中的凈化方法和用量,在均質提取方法下,將得到的提取液分別采用3種凈化填料(NANO- carb、PSA、GCB)的6種配比方案進行凈化(精確到0.5 mg)。具體凈化填料方案為:1)75 mg NANO- carb、125 mg PSA、25 mg GCB;2)75 mg NANO- carb、125 mg PSA;3)45 mg NANO- carb、75 mg PSA、25 mg GCB;4)45 mg NANO- carb、75 mg PSA;5)100 mg NANO- carb、150 mg PSA、25 mg GCB;6)100 mg NANO- carb、150 mg PSA。
6種不同配比的凈化填料方案對于13種農藥的回收率見圖2,各方案RSD均在20%以內。從圖2中可以看出,在方案3的凈化條件下,13種農藥的回收率均在80%~105%,波動較小,總體回收率情況較為優異。此外,在方案4條件下大多數農藥的回收率效果也較好,而且沒有使用GCB,節省了填料;但是在實驗過程中發現,該方案凈化后的提取溶液較方案3顯示出較深的橘黃色,容易造成離子源的污染。綜上,選擇方案3作為優化的凈化填料配比。
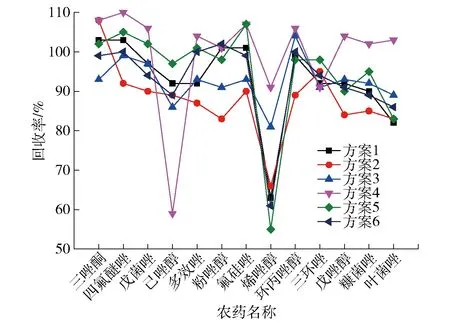
圖2 不同凈化填料配比下13種農藥的回收率Fig.2 Recoveries of 13 pesticides on different conditions of purification fillers
2.4 檢出限、定量限和線性范圍分析
使用空白樣品的綠茶和普洱茶(熟)可以代表未發酵茶和發酵茶。將標準儲備溶液稀釋配制成9個質量濃度梯度的系列混合標準溶液,進行添加驗證。線性范圍、相關系數、方法檢出限(LOD)和定量限(LOQ)見表3和表4。在2~1 000 μg/kg的線性范圍內,綠茶和普洱茶基質中標準曲線的線性關系良好,2種基質的相關系數(R2)均在0.995以上。
測定5份平行樣品,以信噪比(RSN)不低于3計算檢出限,RSN不低于10計算定量限。在2種基質中,13種農藥的檢出限在1~20 μg/kg,定量限在2~50 μg/kg,各自均滿足歐盟MRL要求[19]。
2.5 添加回收驗證分析
進行添加驗證的質量濃度為10、50、100 μg/kg。經檢測和計算,回收率在72%~110%,R2均大于0.995。2種茶葉中13種農藥的添加回收驗證結果見表5和表6。
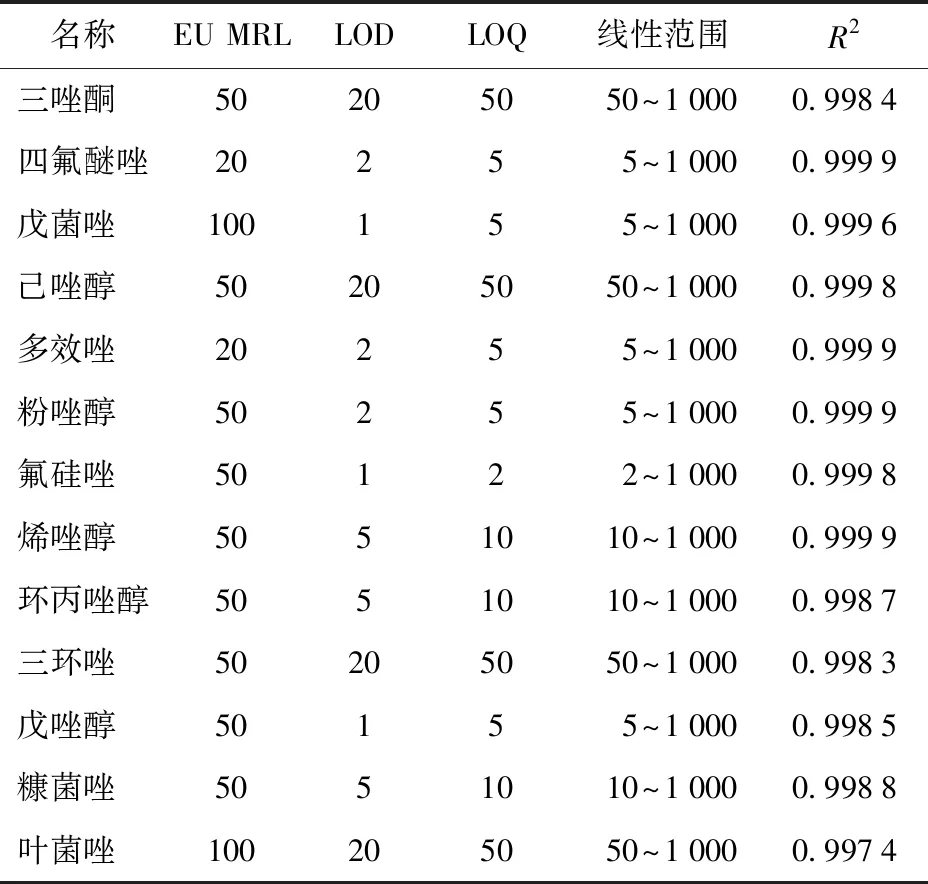
表3 綠茶基質方法學驗證結果Tab.3 Matrix methodology results of green tea μg/kg
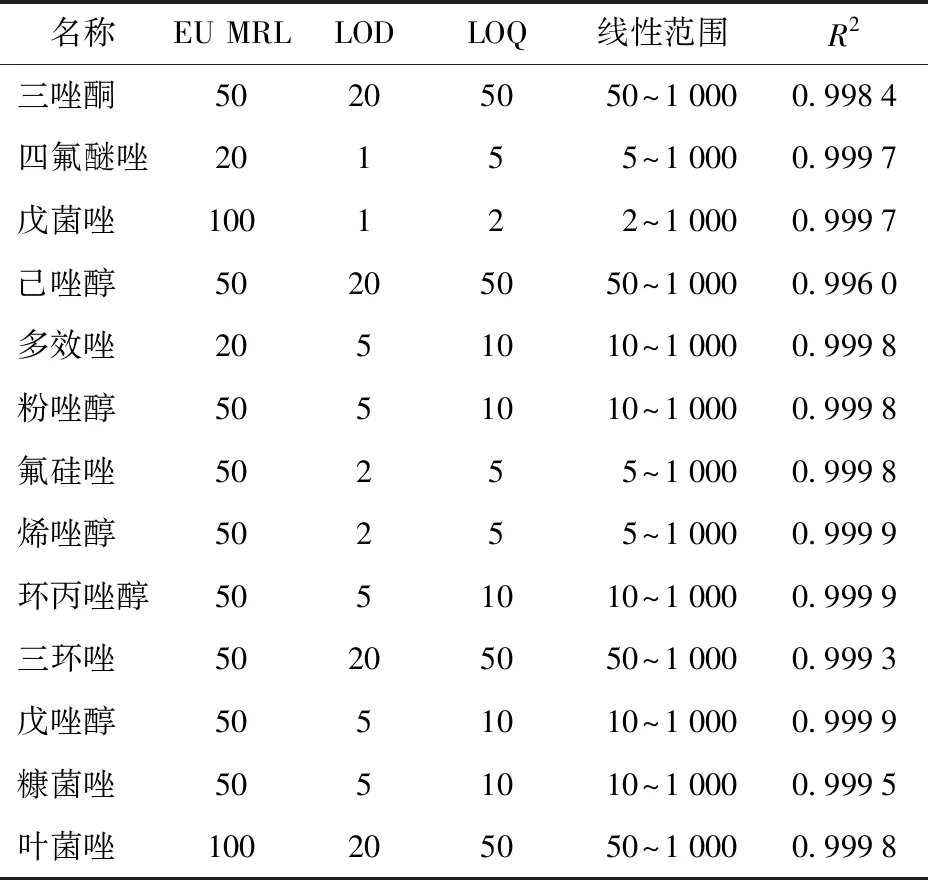
表4 普洱茶基質方法學驗證結果Tab.4 Matrix methodology results of Puer tea μg/kg
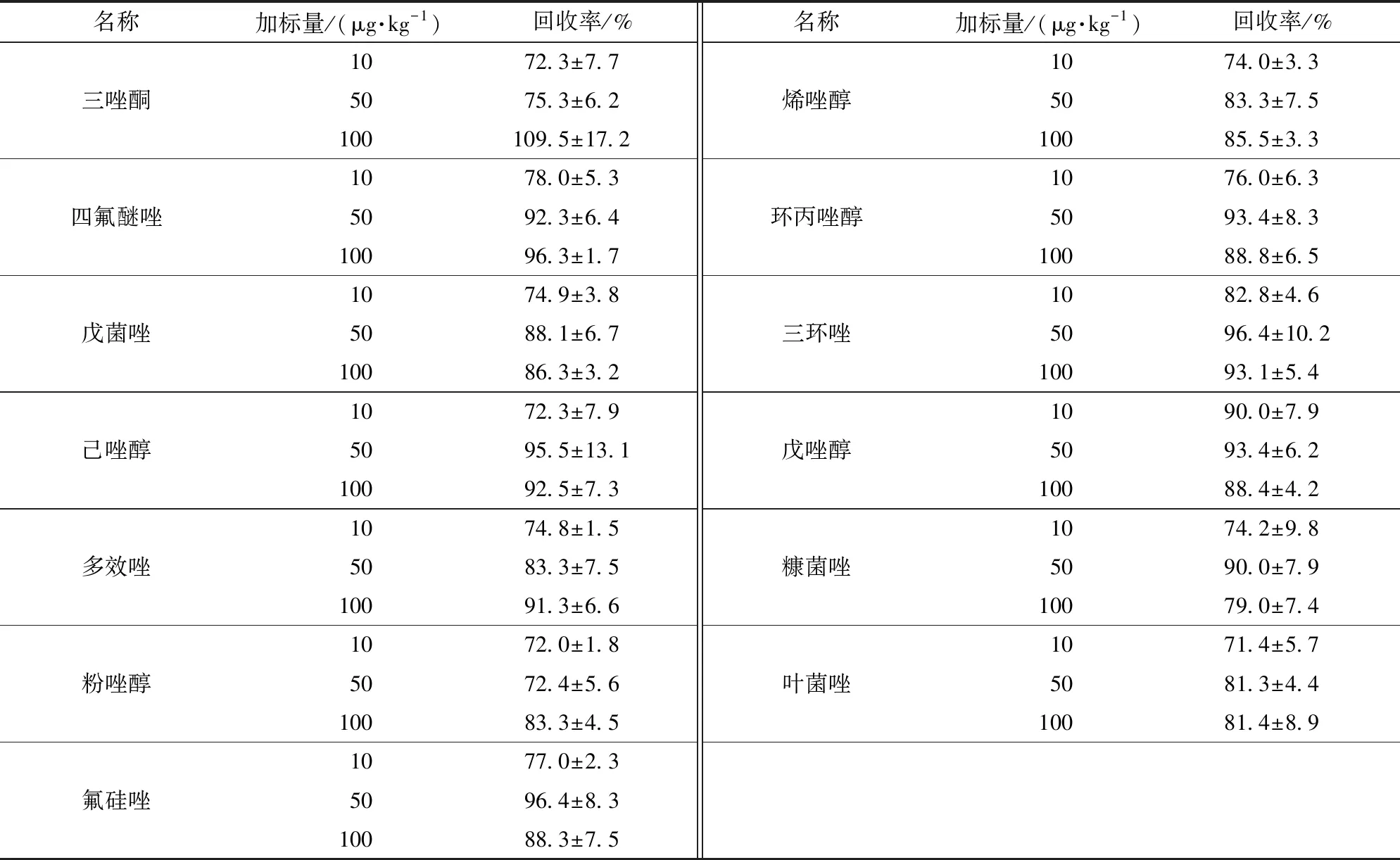
表5 綠茶基質添加回收驗證結果Tab.5 Recovery experiment results of green tea matrix
n=5。
2.6 基質效應的評價
基質效應評價方法為檢測100 μg/kg農藥添加量在茶葉與溶劑中的響應值,兩者差值與后者的比值乘以100%作為評價指標,結果見圖3。數值為正數表示基質增強作用,負數表示減弱作用,±20%以內表示弱基質效應,反之為強基質效應。從圖3中
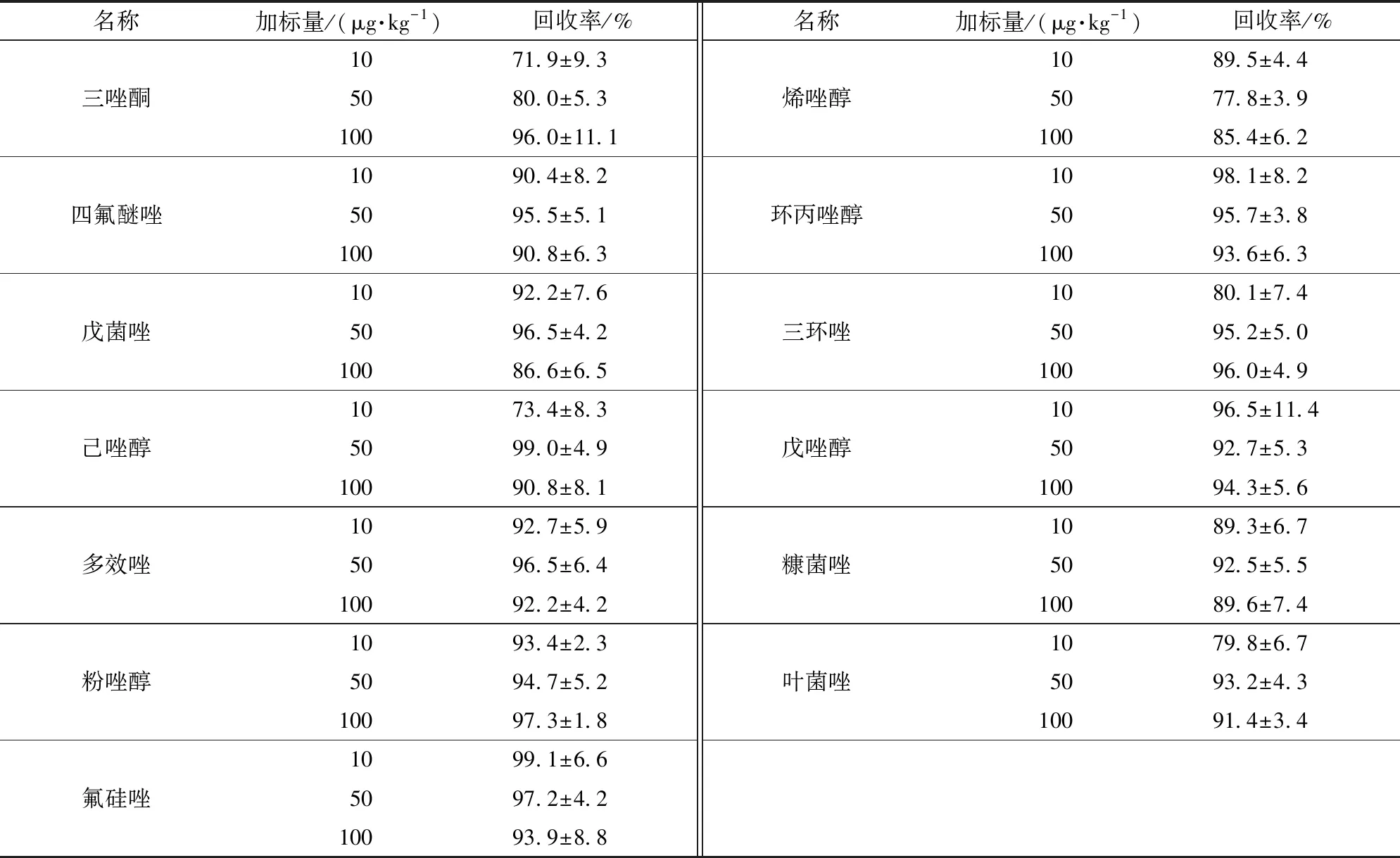
表6 普洱茶基質添加回收驗證結果Tab.6 Recovery experiment results of Puer tea matrix
n=5。
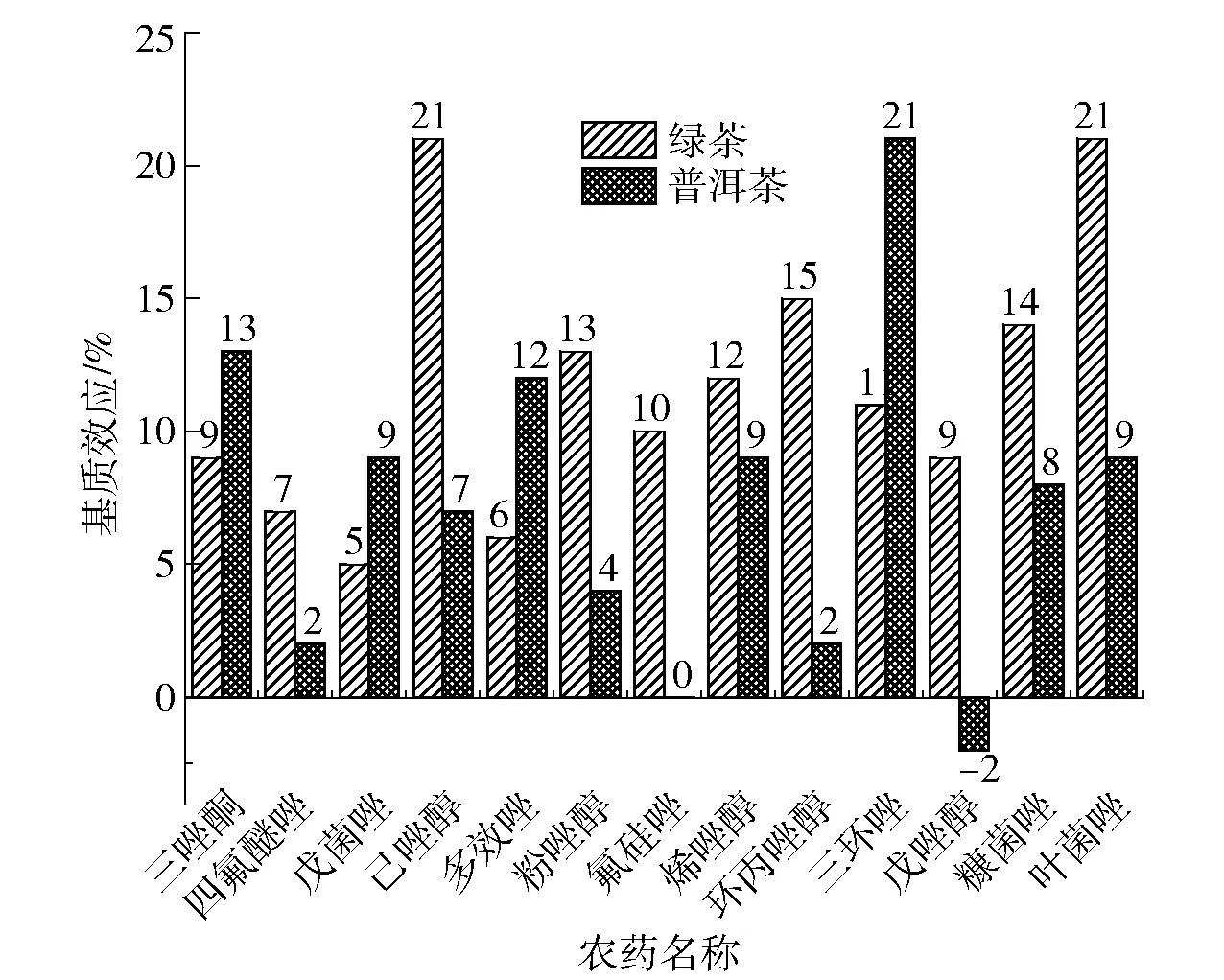
圖3 綠茶和普洱茶的基質效應Fig.3 Matrix effect of green tea and Puer tea
可以看出絕大多數農藥在2種茶葉中呈現基質增強作用,這可能是由于茶葉中的一些成分與檢測系統的活性位點結合彌補了農藥與之結合的損失[20]。綠茶中己唑醇、葉菌唑和普洱茶中三環唑呈現較強的基質效應。而大多數農藥在綠茶中的基質效應較普洱茶稍高,這可能是由于綠茶是未發酵茶,其中的活性成分如色素等造成的干擾更強。
2.7 實際樣品的檢測
使用優化后的前處理方法檢測16例市售綠茶樣品和15例市售普洱茶樣品中13種三唑類農藥含量。2類茶葉中各有1例多效唑檢出:綠茶樣品中檢出的1例含量為31.47 μg/kg,普洱茶中檢測出的1例含量為15.88 μg/kg。
多效唑在歐盟MRL標準中限量為20 μg/kg,則31例樣品中有1例綠茶超標;在日本標準中,依據肯定列表制度—一律標準限量為10 μg/kg,則綠茶和普洱茶各有1例超標。而在國內現行的GB 2763—2016[21]中對于這種主要用作植物生長調節劑的農藥只有其在谷物、油料油脂、蔬菜水果中的最大允許殘留限量,缺少其在茶葉中的限定標準,故無法評價。
3 結 論
研究建立了茶葉中13種三唑類農藥的定性定量檢測方法。樣品采用分散固相萃取凈化,對提取方式和填料配比方案進行對比,使提取效率和凈化效果得到了優化。使用氣相色譜- 四極桿- 飛行時間質譜進行檢測,對色譜升溫條件進行了優化,縮短了檢測時間。在建立的檢測條件下,節省了有機溶劑使用量,方法的回收率良好,檢出限符合歐盟標準的MRL限值,適用于茶葉中的13種三唑類農藥的定性定量分析。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
