金銀花總黃酮自微乳處方的優化
2019-10-24 02:27:06趙惠茹石大玉張一帆
中成藥 2019年10期
關鍵詞:黃酮
趙惠茹,石大玉,靖 會,肖 呈,周 喆,張一帆
(西安醫學院藥學院,陜西 西安 710021)
金銀花為忍冬科植物忍冬Lonicera japonicaThunb.的干燥花蕾或帶初開的花,為臨床常用中藥,具有疏散風熱、清熱解毒的功效[1],其總黃酮有抗氧化、抗炎、抗心律失常、抑菌、保肝等作用[2-3],但其水溶性差,相關口服制劑(金銀花糖漿、復方金銀花顆粒等)生物利用度低,從而限制藥效發揮,影響臨床應用。文獻[4-6]報道,將金銀花總黃酮制成自微乳可能會提高其水溶性,但目前尚無相關研究。
自微乳是由油相、乳化劑、助乳化劑組成的一種新型藥物載體,在溫和攪拌下遇水可自乳化成粒徑小于100 nm 的水包油型乳劑,在增加難溶性藥物溶解度、促進藥物吸收、提高生物利用度、避免藥物水解和對胃腸道不良刺激方面具有明顯優勢,近年來已廣泛用于藥物制劑開發中[7-8]。其中,篩選處方是最核心的部分,故本實驗擬將金銀花總黃酮制成自微乳,并采用單純形網格法優化其處方,為相關新劑型研發提供參考。
1 材料
1.1 儀器 ZEN3600 型激光粒度儀(英國Malvern公司);UV762 型紫外可見分光光度計(上海佑科儀器儀表有限公司);KQ5200E 型超聲波清洗器(昆山市超聲儀器有限公司);FA1004B 型電子精密天平(上海越平科學儀器有限公司);TGL-16C型離心機(上海安亭科學儀器廠);RE-2000A 型旋轉蒸發器、DF-101S 型集熱式恒溫加熱磁力攪拌器(鞏義市予華儀器有限責任公司)。
1.2 試藥 蘆丁對照品(含有量≥99%,批號100080,上海源葉生物科技有限公司);金銀花總黃酮(批號180326,自制,以蘆丁計算含有量為74.6%)。金銀花購于西安市萬壽路藥材市場,經西安醫學院藥學院邊軍昌高級實驗師鑒定為忍冬科植物忍冬Lonicera japonicaThunb.的干燥花蕾。D101 大孔吸附樹脂(陜西藍深科技有限公司);聚氧乙烯氫化蓖麻油(RH-40)、聚氧乙烯蓖麻油(EL-35)(德國巴斯夫公司);曲拉通(X-100,成都市科龍化工試劑廠);吐溫-80(天津富宇精細化工有限公司);辛酸癸酸三甘油酯(上海千為油脂科技有限公司);肉豆蔻酸異丙酯(國藥集團化學試劑有限公司);油酸乙酯(天津市光復精細化工研究所);聚乙二醇400(天津市科密歐化學試劑有限公司);1,2-丙二醇(天津市北聯精細化學品開發有限公司);異丙醇(天津市河東區紅巖試劑廠)。其他試劑均為分析純;水為超純水。
2 方法與結果
2.1 總黃酮含有量測定[9]精密稱取蘆丁對照品20.0 mg,置于50 mL 量瓶中,70%乙醇溶解定容,得0.400 mg/mL 對照品溶液,精密移取0.0、1.0、1.5、2.0、2.5、3.0、3.5、4.0 mL,置于10 mL量瓶中,依次加入5%亞硝酸鈉0.4 mL,搖勻,靜置6 min;10%硝酸鋁0.4 mL,搖勻,靜置6 min;4%氫氧化鈉4 mL,加水定容至刻度,搖勻,靜置15 min,以相應試劑為空白對照,在510 nm 波長處測定吸光度。以溶液質量濃度為橫坐標(X),吸光度為縱坐標(A)進行回歸,得方程為A=4.366 1X+0.011 3(r=0.997 1),在0.04~0.16 mg/mL范圍內線性關系良好。
2.2 總黃酮制備
2.2.1 提取[10]稱取藥材粗粉適量,置于索氏提取器中,石油醚連續回流至提取液無色后取出晾干,加入10、8、6 倍量70% 乙醇回流提取3 次,每次1.5 h,提取液合并,減壓濃縮至無醇味,6倍量蒸餾水攪拌溶解,12 h 后離心,上清液減壓濃縮至適當濃度,備用。
2.2.2 純化[11-12]將預處理好的D101 大孔吸附樹脂倒入燒杯中,加入總黃酮濃縮液靜態吸附12 h,將達到吸附飽和的樹脂裝入層析柱中,依次用6 BV 蒸餾水、5 BV 30%乙醇洗脫除去雜質,再用5 BV 70%乙醇洗脫,收集洗脫液,減壓回收乙醇,即得總黃酮。精密稱取干燥至恒重的總黃酮50 mg,置于25 mL 量瓶中,70%乙醇溶解定,精密移取2.0 mL 置于25 mL 量瓶中,按“2.1”項下方法測定吸光度,測得總黃酮含有量為74.6%。
2.3 處方篩選
2.3.1 溶解度 精密稱取聚氧乙烯氫化蓖麻油RH-40、曲拉通X-100、聚氧乙烯蓖麻油EL-35、吐溫-80 這4 種乳化劑,以及異丙醇、聚乙二醇400、1,2-丙二醇、無水乙醇這4 種助乳化劑各4 g,置于具塞離心管中,加入過量總黃酮,在20 kHz頻率、室溫下超聲1 h 輔助溶解后37 ℃水浴中靜置24 h,3 000 r/min 離心10 min,吸取適量上清液,甲醇稀釋至適宜濃度,按“2.1”項下方法測定吸光度,計算溶解度,結果見表1。由表可知,總黃酮在聚氧乙烯氫化蓖麻油RH-40、吐溫-80、無水乙醇、1,2-丙二醇中的溶解度較大,故選擇前兩者作為乳化劑,后兩者作為助乳化劑進一步篩選自微乳油相。

表1 總黃酮溶解度測定結果Tab.1 Results of solubility determination of total flavonoids
2.3.2 油相與乳化劑配伍 將辛酸癸酸三甘油酯、肉豆蔻酸異丙酯、油酸乙酯這3 種油相與聚氧乙烯氫化蓖麻油RH40、吐溫-80 這2 種乳化劑按1∶9、2∶8、3∶7、4∶6 比例混合后,加入100 倍量超純水,400 r/min 磁力攪拌,按文獻[13]方法根據乳化時間和液體顏色觀察乳化情況。結果,油酸乙酯與吐溫-80 配伍時乳化時間短,所需乳化劑少,乳化效果好,故選擇前者作為油相,后者作為乳化劑。
2.3.3 助乳化劑 按照油酸乙酯、吐溫-80、助乳化劑2∶4∶4 的比例加入助乳化劑無水乙醇、1,2-丙二醇,400 r/min 磁力攪拌均勻后加入100 倍量超純水磁力攪拌乳化,記錄從加入超純水開始磁力攪拌至完全分散、形成外觀澄清或有藍白色乳光均一微乳所需的時間,即為乳化時間。結果,加入2 種助乳化劑后乳化時間均分別縮短至24、37 s,考慮到無水乙醇易揮發,故選擇1,2-丙二醇作為助乳化劑。
2.3.4 各組分比例范圍[14]先將吐溫-80 與1,2-丙二醇按照1∶9、2∶8、3∶7、4∶6、5∶5、6∶4、7∶3、8∶2、9∶1 比例混勻,再與油酸乙酯按照9∶1、8∶2、7∶3、6∶4、5∶5 比例混勻后,稱取0.5 g 滴加到50 mL 超純水中,400 r/min 磁力攪拌,記錄可形成外觀澄清或有藍白色乳光的均一微乳處方中各組分比例。再以油相、乳化劑、助乳化劑為相圖3 個頂點,Origin8.0 軟件繪制偽三元相圖,結果見圖1,根據微乳區域的邊界確定油酸乙酯、吐溫-80、1,2-丙二醇比例范圍分別為10%~30%、30%~70%、10%~50%。
2.4 處方優化
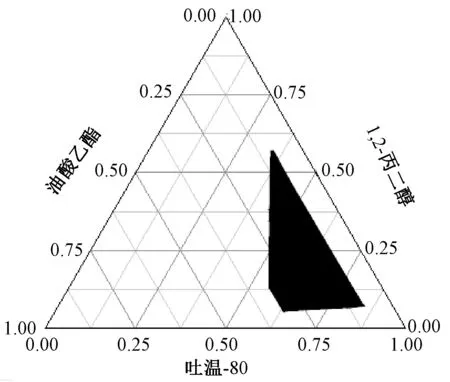
圖1 自微乳偽三元相圖Fig.1 Pseudo-ternary phase diagram for self-microemulsions
2.4.1 試驗設計[15-17]根據“2.3”項下處方篩選結果,結合單純形網格法比例約束,確定自微乳中各組分比例為油酸乙酯(X1)10%~25%、吐溫-80(X2)45%~60%、1,2-丙 二 醇(X3)30%~45%。再以自微乳平均粒徑(Y1)、多分散指數(Y2)、載藥量(Y3)為評價指標,Design Expert 8.0.6 軟件進行設計,按處方比例精密稱取油相、乳化劑、助乳化劑,400 r/min 磁力攪拌10 min后加入過量總黃酮,混勻,在20 kHz 頻率、室溫下超聲1 h 輔助溶解,靜置24 h,即得自微乳。過濾后,精密稱取0.5 g 置于50 mL 量瓶中,超純水稀釋并定容,搖勻,3 000 r/min 離心10 min,移取適量上清液,測定平均粒徑、多分散指數,按“2.1”項下方法測定吸光度,計算載藥量,公式為載藥量=總黃酮質量/自微乳質量,結果見表2。
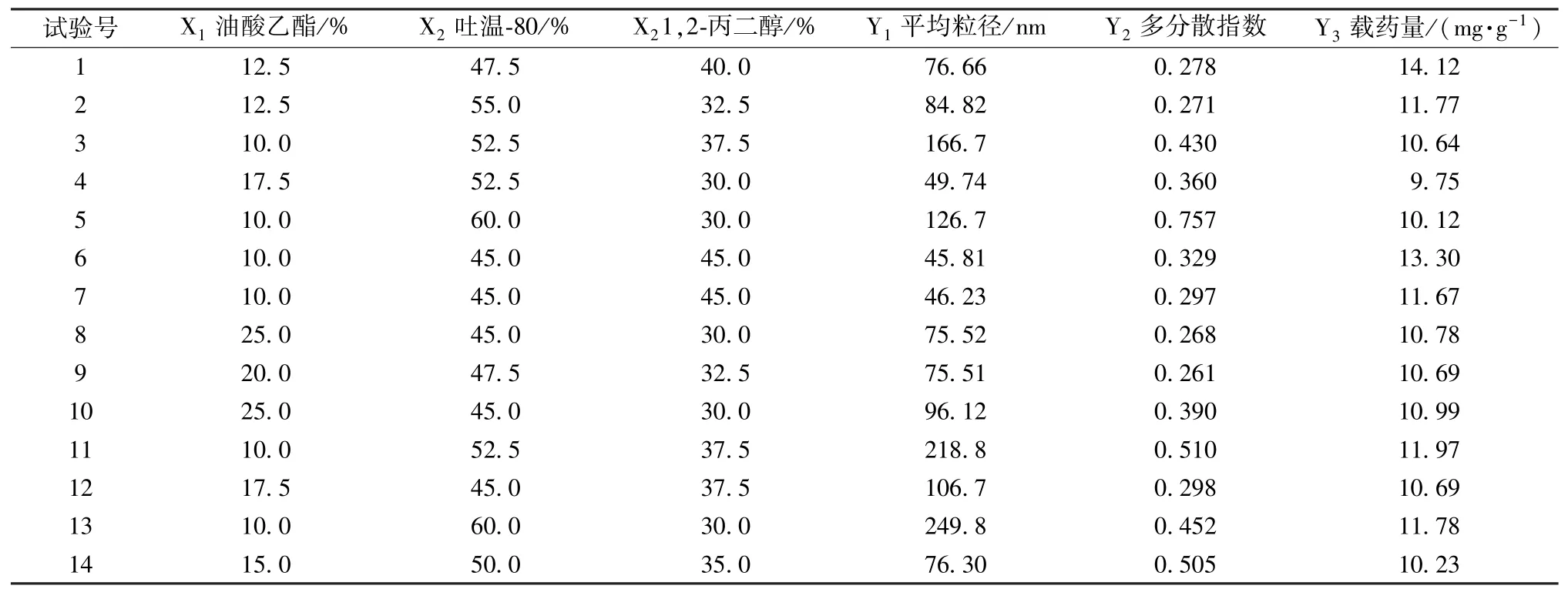
表2 試驗設計及結果Tab.2 Design and results of tests
2.4.2 模型擬合及方差分析 通過Design Expert 8.0.6 軟件進行多元回歸模型擬合,得方程分別為Y1=-77.772 60X1-77.945 24X2-19.639 91X3+11.678 00X1X2+5.708 27X2X3+20.655 94X1X3-0.441 59X1X2X3、Y2=-0.005 48X1+0.012 17X2-0.004 11X3、Y3=-0.051 34X1-0.072 01X2+0.197 74X3。再進行方差分析,可知各模型P<0.000 1,失 擬 項P>0.05,回 歸 方 程 系 數R2>0.80,表明模型擬合度良好。
2.4.3 響應面分析 見圖2。
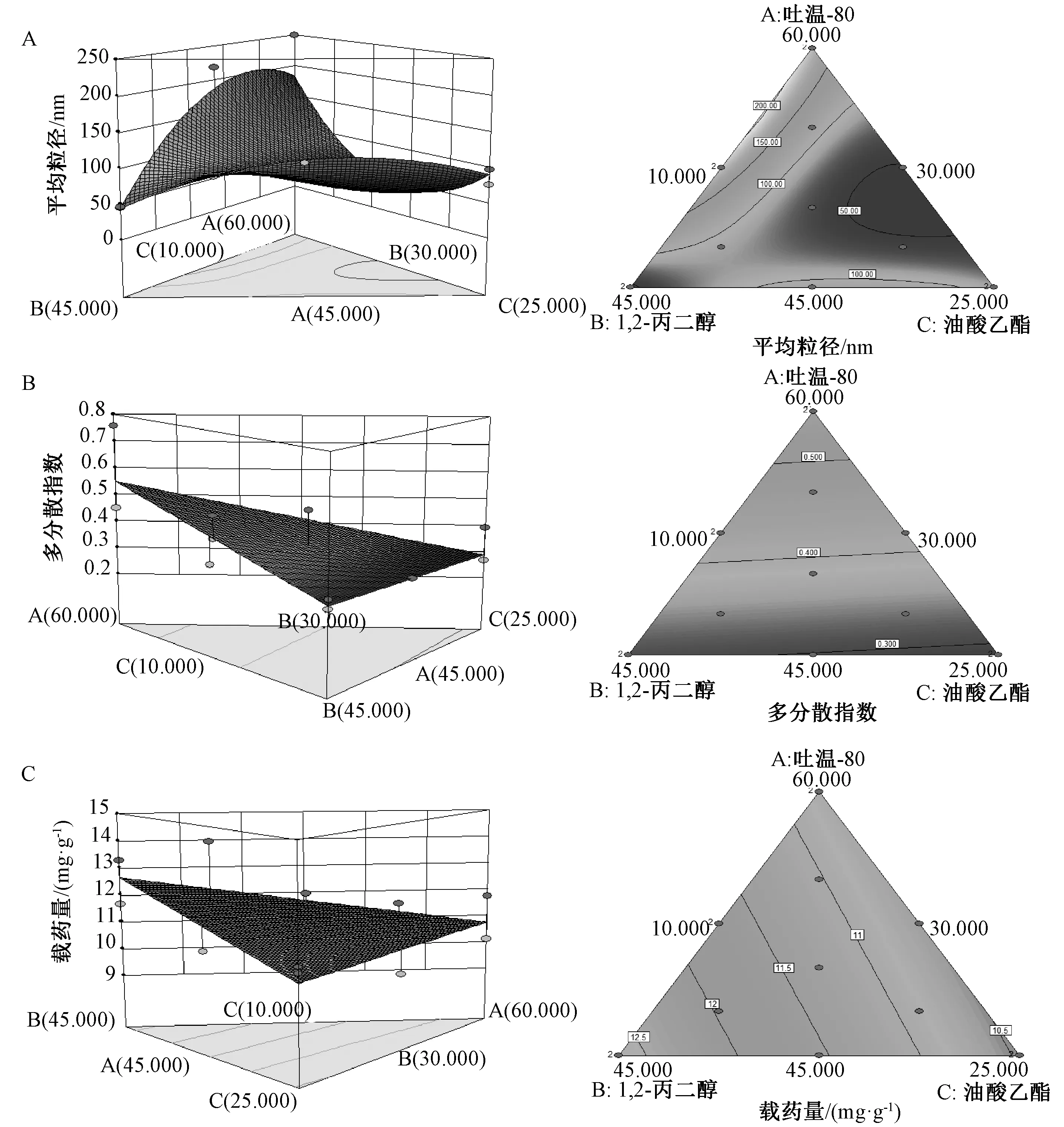
圖2 各因素響應面圖Fig.2 Response surface plots for various factors
2.5 驗證試驗 自微乳粒徑越小,粒徑分布越窄,越有利于藥物的溶出與吸收;載藥量越大,實際服用劑量越少,越便于臨床應用。因此,綜合考慮3個指標,以平均粒徑、多分散指數符合要求并盡可能小,載藥量最大為指標,Design Expert 8.0.6 軟件進行優化,得最優處方為油酸乙酯、吐溫-80、1,2-丙二醇比例10∶45∶45。再根據優化處方制備3 批自微乳,進行驗證試驗,結果見表3、圖3,可知處方穩定可行。
3 討論

圖3 自微乳粒徑分布Fig.3 Particle size distribution of self-microemulsions
本實驗采用D101 大孔吸附樹脂分離純化金銀花總黃酮時,依次用適量純化水和30%乙醇除雜,再用70%乙醇洗脫,發現純度較高,粒徑較預實驗沒有除雜所得的自微乳減小,載藥量顯著增大,說明理化性質差別較大的雜質會對其形成產生特殊影響。因此,在對中藥有效部位自微乳處方進行研究時,應盡可能采用多種純化方法提高其純度。
本實驗首先根據金銀花總黃酮在幾種備選乳化劑、助乳化劑中的溶解度初步篩選,然后通過油相、乳化劑的配伍實驗來確定種類,接著篩選助乳化劑,根據偽三元相圖確定各自比例范圍,最后用單純形網格法進一步優化。這與測定總黃酮在各種備選油相、乳化劑、助乳化劑中的溶解度來篩選處方不同,主要是因為即使總黃酮在某種油相中的溶解度最大,與乳化劑、助乳化劑配伍后未必是最優處方,甚至有時候并不能形成穩定的微乳,還需要進一步選擇溶解度次之的油相去配伍。為了減少油相、乳化劑、助乳化劑配伍次數,避免盲目性篩選,本實驗略去了總黃酮在各種備選油相中的溶解度數據。
由于金銀花總黃酮在無水乙醇中的溶解度大于1,2-丙二醇,故預實驗以其為助乳化劑,偽三元相圖微乳區域各組分的比例分別為10%~40%油酸乙酯、30%~80% 吐溫-80、10%~60% 無水乙醇,但用單純形網格法優選時有個別處方不能形成穩定的微乳,可能是由于攪拌時無水乙醇揮發,從而使各組分比例發生變化,降低了穩定性,故選用對金銀花總黃酮溶解能力較好、不揮發的1,2-丙二醇作為助乳化劑。另外,采用單純形網格法優選處方時,14 組實驗形成的空白微乳均為澄清或略泛藍色,并均能在室溫下穩定存在。
處方篩選優化是自微乳制劑研究最關鍵的部分,目前可應用正交試驗、均勻設計實驗、星點設計-效應面法等進行研究,每種方法都有其優缺點。其中,基于Design Expert 8.0.6 軟件試驗設計、模型構建、結果預測的單純形網格法是采用空間圖形淘汰法進行處方優選,通過因素之間的相互約束來影響指標,在確定處方組成的響應面和最佳范圍方面優勢獨到,尤其適合自微乳這類組分總量恒定的劑型,而且考察因素數目相同時實驗次數少于星點設計-效應面法,不必連續進行多次試驗,預測結果準確。綜上所述,單純形網格法拓寬了自微乳處方篩選方法,可為相關優化研究提供新思路。
猜你喜歡
四川蠶業(2021年2期)2021-03-09 03:15:32
四川蠶業(2021年3期)2021-02-12 02:38:46
中成藥(2018年11期)2018-11-24 02:57:00
中成藥(2017年8期)2017-11-22 03:19:40
中成藥(2017年10期)2017-11-16 00:50:13
中成藥(2017年4期)2017-05-17 06:09:50
哈爾濱醫藥(2016年1期)2017-01-15 13:43:16
天然產物研究與開發(2016年11期)2016-06-15 20:29:17
湖南師范大學自然科學學報(2015年1期)2015-02-27 14:50:04
安徽醫藥(2014年12期)2014-03-20 13:15:15
