親水作用色譜-串聯質譜法測定動物源食品中10種氨基糖苷類藥物的殘留量
2019-10-12 02:50:02宓捷波張敏柴銘駿劉旸常春艷
食品研究與開發 2019年19期
宓捷波,張敏,柴銘駿,劉旸,常春艷
(天津出入境檢驗檢疫局動植物與食品檢測中心,天津300461)
氨基糖苷類藥物是由鏈霉菌或小單胞菌培養液中提取,或合成的一類水溶性堿性抗生素,可作用于細菌體內的核糖體,抑制細菌蛋白質的合成,并破壞細菌細胞膜的完整性,對需氧革蘭氏陰性菌、單胞菌屬、葡萄球菌屬及結核桿菌均有抗菌活性[1-3]。氨基糖苷類藥物核心的分子結構為一個氨基環醇環和一個或多個氨基糖分子通過配糖鍵相連接,典型的代表藥物有鏈霉素、雙氫鏈霉素、新霉素、卡那霉素、慶大霉素等。在農業、畜牧業和水產業中,氨基糖苷類藥物可以有效抑制細菌的生長和繁殖,因此常被作為獸藥治療家畜腸炎、赤皮病、白頭白嘴病等。由于對藥物的不合理使用及違法使用等情況,導致的氨基糖苷類抗生素在食品中殘留問題,已經引起國內外的普遍關注。如美國、加拿大等國相關機構調查發現鏈霉素是僅次于青霉素的最常在動物中殘留超標的藥物;國內也曾在豬腎豬肝中檢出嚴重超標的鏈霉素。醫學的進一步研究也發現,氨基糖苷類藥物雖然具有強大的殺菌作用,但會導致人體內蛋白質合成異常,從而引發口周和手足麻木、神經性的肌肉阻滯,蛋白尿、腎小球濾過減少、氮質血癥等腎臟疾病。因此,有必要對動物源食品中的氨基糖苷類藥物殘留進行檢測。
氨基糖苷類藥物的檢測本身并不困難,如微生物法[4]、酶聯免疫法[5-8]、液相色譜法[9-12]、氣相色譜法[13]、液相色譜-串聯質譜法[14-19]等都曾用于食品、生物體液及環境中氨基糖苷類藥物的檢測。對于食品中氨基糖苷類藥物的殘留分析而言,液相色譜-串聯質譜法以其特有的定性定量準確的特征成為近年來食品行業中的重要方法。由于氨基糖苷類藥物極性大,在常規反相色譜柱上保留性能不佳,往往采用在流動相中加入烷基磺酸鈉或七氟丁酸等離子對試劑改善保留性能。然而,離子對試劑對質譜性能會造成不可逆轉的影響[20],導致質譜對負離子的分析性能降低,即分析氨基糖苷類藥物用的質譜將只能用于正離子測定,這也是目前該類藥物分析面臨的最大困難。因此,不采用離子對試劑作為流動相組分的親水作用色譜(hydrophilic interaction chromatography,HILIC)逐漸成為氨基糖苷類等極性物質分析研究的熱點。本研究通過對不同鍵合相的親水作用色譜條件進行優化,建立動物源性食品中10 種氨基糖苷類藥物殘留量的檢測方法,克服常規方法損傷質譜的缺陷,提高方法的實用性。
1 材料與方法
1.1 儀器與試劑
1.1.1 材料與試劑
試驗用的豬肉、牛肉、豬腎和牛肝均為天津口岸進出口的日常檢測樣品。
乙腈、甲酸(色譜純):德國Merck 公司;三氯乙酸、七氟丁酸、庚烷磺酸鈉(分析純):Sigma-Aldrich 公司(美國);乙二胺四乙酸二鈉(ethylenediaminetetraacetate dehydrate,EDTA-2Na)、磷酸、鹽酸、氫氧化鈉、磷酸二氫鉀(分析純):天津試劑公司;丁胺卡那霉素、潮霉素B、奇霉素、鏈霉素、雙氫鏈霉素、卡那霉素、新霉素、安普霉素、妥布霉素和慶大霉素(純度大于99%):Dr.E hretorfer GmbH 公司。試驗用水為去離子水。
1.1.2 儀器與設備
Agilent 1200-API 4000 液相色譜-串聯質譜儀:AB Sciex 公司;Sielc Obelisc R 色譜柱(150 mm×2.1 mm,5 μm):飛諾美公司;ACQUITY UPLC BEH HILIC 色譜柱(100 mm×2.1 mm,1.7 μm):沃特世公司;GL Sciences InertSustain Amide 色譜柱(100 mm×2.1 mm,3 μm):島津(上海)試驗器材有限公司;TSKgel Amide-80 色譜柱(150 mm×2.0 mm,5 μm):東曹(上海)生物科技有限公司;IKA HS501 水平往復振蕩儀、IKA MS3 basic 渦旋混合器:艾卡(廣州)儀器設備有限公司;Avanti J-30I高速冷凍離心機:貝克曼庫爾特商貿(中國)有限公司;SPE-24 固相萃取裝置:博納艾杰爾科技有限公司;NEVAP-34 氮氣濃縮儀:organomation 公司。
1.2 樣品預處理
稱取試樣5 g(精確至0.01 g)于50 mL 離心管中,加入10 mL 含0.4 mmol/L EDTA、2%三氯乙酸的磷酸緩沖液(pH 4.0),振蕩提取10 min,離心取上清,對殘渣重復提取1 次,合并兩次提取的上清液,用1 mol/L鹽酸調pH 值為3.5,加入2 mL 100 mmol/L 七氟丁酸溶液,渦旋混勻后在預先經3 mL 甲醇、3 mL 去離子水活化的C18 固相萃取柱上樣,然后先用3 mL 20 mmol/L七氟丁酸溶液洗滌,再用水洗滌2 次,每次3 mL,抽干,最后用5 mL 乙腈-20 mmol/L 七氟丁酸溶液(體積比 8 ∶2)洗脫,洗脫液于40 ℃氮吹至小于1 mL,用含4 %甲酸的0.01 mol/L 庚烷磺酸鈉溶液定容至1 mL,過膜,待測。
1.3 色譜質譜條件
色譜條件:Sielc Obelisc R 色譜柱(150 mm×2.1 mm,3.5 μm),流速 0.3 mL/min,進樣量 20 μL,流動相 A 為1%的甲酸水,流動相B 為乙腈,流動相C 為水,采用梯度進行洗脫分離。
梯度洗脫程序如表1 所示。
質譜條件:采用正離子掃描模式,噴霧電壓為5 500 V,霧化氣壓力為12 psi(1 psi=0.006 89 MPa),氣簾氣壓力為20 psi,輔助氣1 壓力為55 psi,輔助氣2壓力為50 psi,離子源溫度為600 ℃,采用多反應監測模式。
10 種氨基糖苷類藥物的定性定量離子對等信息見表2。
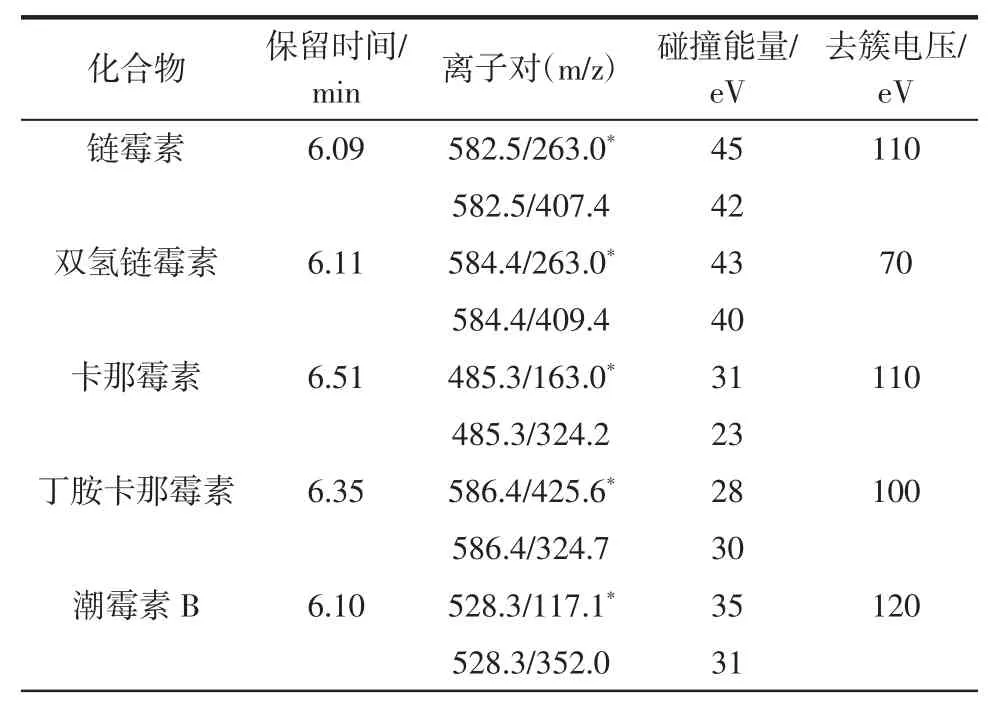
表2 10 種氨基糖苷類藥物的質譜分析參數Table 2 MS paramaters for 10 aminoglycosides
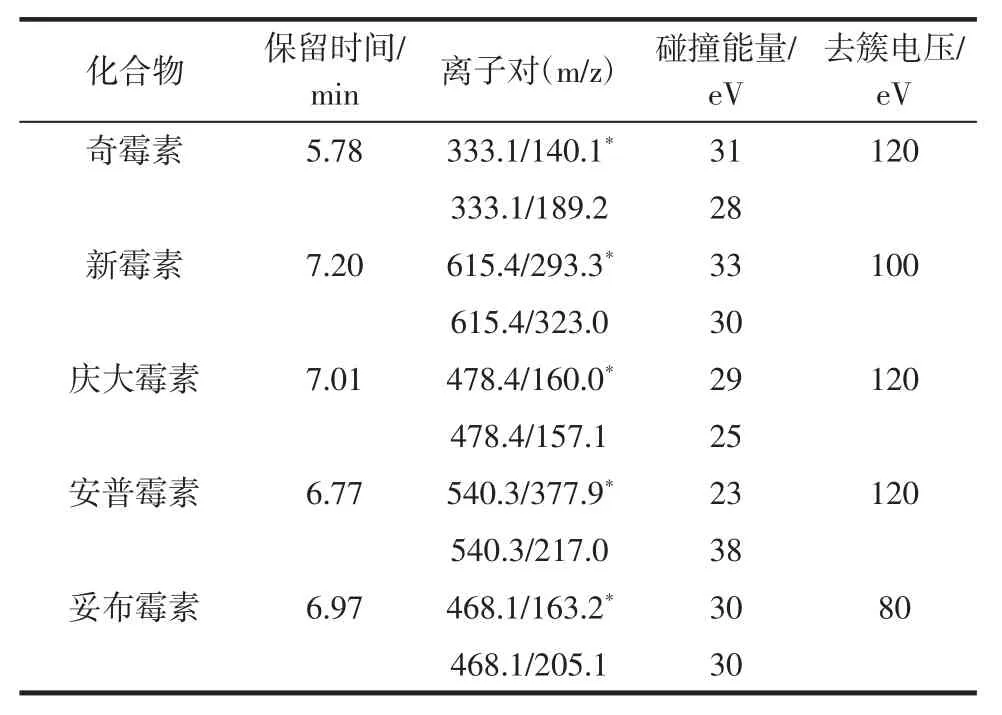
續表2 10 種氨基糖苷類藥物的質譜分析參數Continue table 2 MS paramaters for 10 aminoglycosides
2 結果與分析
2.1 親水作用色譜柱的選擇
一般認為,親水作用色譜的保留機制主要包括鍵合固定相水層和流動相之間的分配作用、氫鍵或偶極之間的吸附作用以及靜電作用[21-22]。親水作用色譜的鍵合固定相種類繁多,包括未衍生的硅膠以及氨基、酰胺基、氰基修飾的硅膠等[23]。不同的鍵合固定相在分離目標物的過程中往往保留機制迥異,需要通過具體的試驗進行選擇。本試驗主要對ACQUITY UPLC BEH HILIC 柱(鍵合相為硅膠)、GL Sciences InertSustain Amide(鍵合相為酰胺基)柱、TSKgel Amide-80(鍵合相為酰胺基)柱和Sielc Obelisc R(鍵合相為含電荷的極性和非極性混合基團)色譜柱分離氨基糖苷類藥物的效果進行了比較。
潮霉素B、安普霉素和新霉素在3 種色譜柱上的分離色譜圖見圖1。
試驗結果顯示,BEH HILIC 柱分離氨基糖苷類藥物效果不佳,其余3 種色譜柱可以對目標物實現分離。其中InertSustain Amide 柱在流動相含100 mmol/L 甲酸銨的條件下以乙腈和水可以實現氨基糖胺類物質的分離,但潮霉素B、奇霉素會出現雙峰,目標物響應強度相應較弱。而TSK gelAmide-80 柱和Sielc Obelisc R 鍵合相的親水作用色譜柱對目標物的分離較為相似,采用1%甲酸水和乙腈為流動相的條件下可以實現氨基糖苷類藥物的分離。TSKgel Amide-80 柱上目標物質雖為單一峰形,但峰形有展寬,且拖尾,而含有陽離子極性與非極性混合作用機理模式的Sielc O-belisc R 色譜柱上各物質的分離效果最好。故最終采用Sielc Obelisc R 色譜柱。
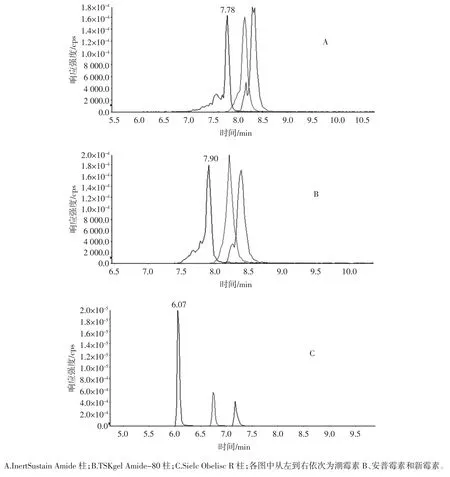
圖1 潮霉素B、安普霉素和新霉素在3 種色譜柱上的分離圖Fig.1 Separation of hygromycin B,apramycin and neomycin in different column
2.2 流動相pH值的優化
氨基糖苷類藥物包含多個氨基結構,屬于堿性糖苷,因此在與Sielc Obelisc R 柱填料相互作用的過程中,流動相的pH 值會顯著影響氨基糖苷類物質的電荷狀態,進而影響其在色譜柱上受到的靜電作用的大小,導致分離性能出現差異[24]。本研究分別比較了0.1%甲酸水,0.5 %甲酸水和1 %甲酸水對于目標物分離的影響。
妥布霉素在不同甲酸含量流動相中的分離情況見圖2。
由圖2 可知,1%甲酸水作為流動相的效果最佳。這是由于流動相的pH 值越低,堿性的氨基糖苷類藥物Sielc Obelisc R 柱填料表面均帶正電荷,存在排斥作用,避免了因靜電吸引而導致的峰形拖尾,同時保留時間也明顯縮短。試驗中,InertSustain Amide 柱和TSKgel Amide-80 柱在1%甲酸水的條件下拖尾現象也有所改善,但峰整體展寬很嚴重,說明酰胺基的鍵合相對目標物的吸附作用更強,不易洗脫。
2.3 HILIC色譜柱分析穩定條件的探索
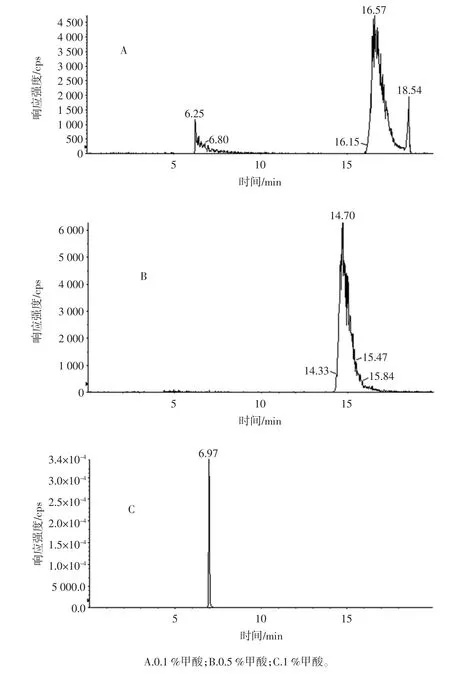
圖2 流動相pH 值對親水作用色譜分離氨基糖苷類藥物的影響Fig.2 The effect of pH value of mobile phase during the separation of aminoglycosides using HILIC column
一些文獻[21-25]顯示,HILIC 機制的色譜柱在使用過程中往往存在保留時間不穩定的情況,本研究也對氨基糖苷類藥物在HILIC 柱上分析的穩定性進行了研究。結果顯示,HILIC 柱需要比常規反相色譜柱更長的平衡時間,一旦流動相條件發生變化,至少需要進行30 min 以上的重新平衡時間。而且梯度洗脫程序中的平衡時間也相對較長,本方法在目標物洗脫完成后10 min 才使柱壓恢復到起始壓力。此外,HILIC 柱需保持高比例的有機相,因此每次試驗后需及時將色譜柱從儀器上取下,否則易出現HILIC 柱中有機相揮發導致的柱壓上升并進而影響分離的情況。鑒于上述原因,本方法的梯度洗脫程序后期采用了近11 min 的平衡時間,且保證每次使用后及時將色譜柱兩端封口。在這種條件下,HILIC 柱對氨基糖苷類藥物的分離效果十分穩定。
2.4 凈化柱和定容溶液的優化
盡管HILIC 填料固相萃取(solid phase extraction,SPE)柱或定制柱也可用于氨基糖苷類藥物的凈化,但商品化產品相對較少,定制價格較高,且洗脫條件隨目標物的類別變化而變化,通用性較差,目前國內外應用相對較少。鑒于氨基糖苷類藥物強極性和堿性的特征,離子交換柱和C18 柱(配合離子對試劑)SPE 是文獻中應用較多的兩種固相萃取柱。本研究中分別用弱陽離子交換柱(weak cation exchange,WCX)和C18 SPE 柱對豬肉組織中10 種氨基糖苷類藥物進行凈化,結果顯示鏈霉素、雙氫鏈霉素和卡那霉素用WCX 柱凈化時效果較好,但其他7 種物質的凈化效果不理想,而C18 SPE 柱在七氟丁酸離子對和酸性緩沖液的作用下對10 種氨基糖苷類藥物均能實現較好的凈化效果。這可能與氨基糖苷類藥物本身帶電荷狀態受pH 值影響有關,離子交換作用可能導致部分物質與填料發生較強的作用而不易洗脫,而離子對試劑存在下,目標物在C18 SPE 柱上僅受極性作用,相對洗脫較為順利。因此,本研究最終采用C18 SPE 柱配合七氟丁酸溶液進行凈化處理。
對于定容溶液的選擇,試驗中比較了七氟丁酸和庚烷磺酸鈉溶液,結果顯示庚烷磺酸鈉定容后氨基糖苷類藥物的色譜分離效果更好。
2.5 方法的標準曲線和定量限
混合標準工作曲線采用空白豬肉基質提取液稀釋混合標準儲備溶液的方式配制,再以親水作用色譜串聯質譜檢測,計算擬合標準曲線。試驗結果顯示,以測定結果的峰面積(y)為縱坐標,分析物的濃度(x)為橫坐標,10 種氨基糖苷類藥物在50 ng/mL~5 000 ng/mL范圍內線性良好,相關系數均大于0.99。同時,通過在空白樣品中進行加標測定,以提取離子色譜峰的信噪比(S/N)大于10 計算方法的定量限[26]:新霉素、潮霉素B 和安普霉素為100 μg/kg,奇霉素、丁胺卡那霉素、鏈霉素、雙氫鏈霉素、卡那霉素、慶大霉素和妥布霉素為20 μg/kg,可以滿足目前國內外對氨基糖苷類藥物的最大殘留限量的要求。
10 種氨基糖苷類物質的提取離子色譜圖見圖3。
由圖3 可以看出,10 種氨基糖苷類藥物可在本研究條件下獲得很好的分離,并且峰形尖銳。
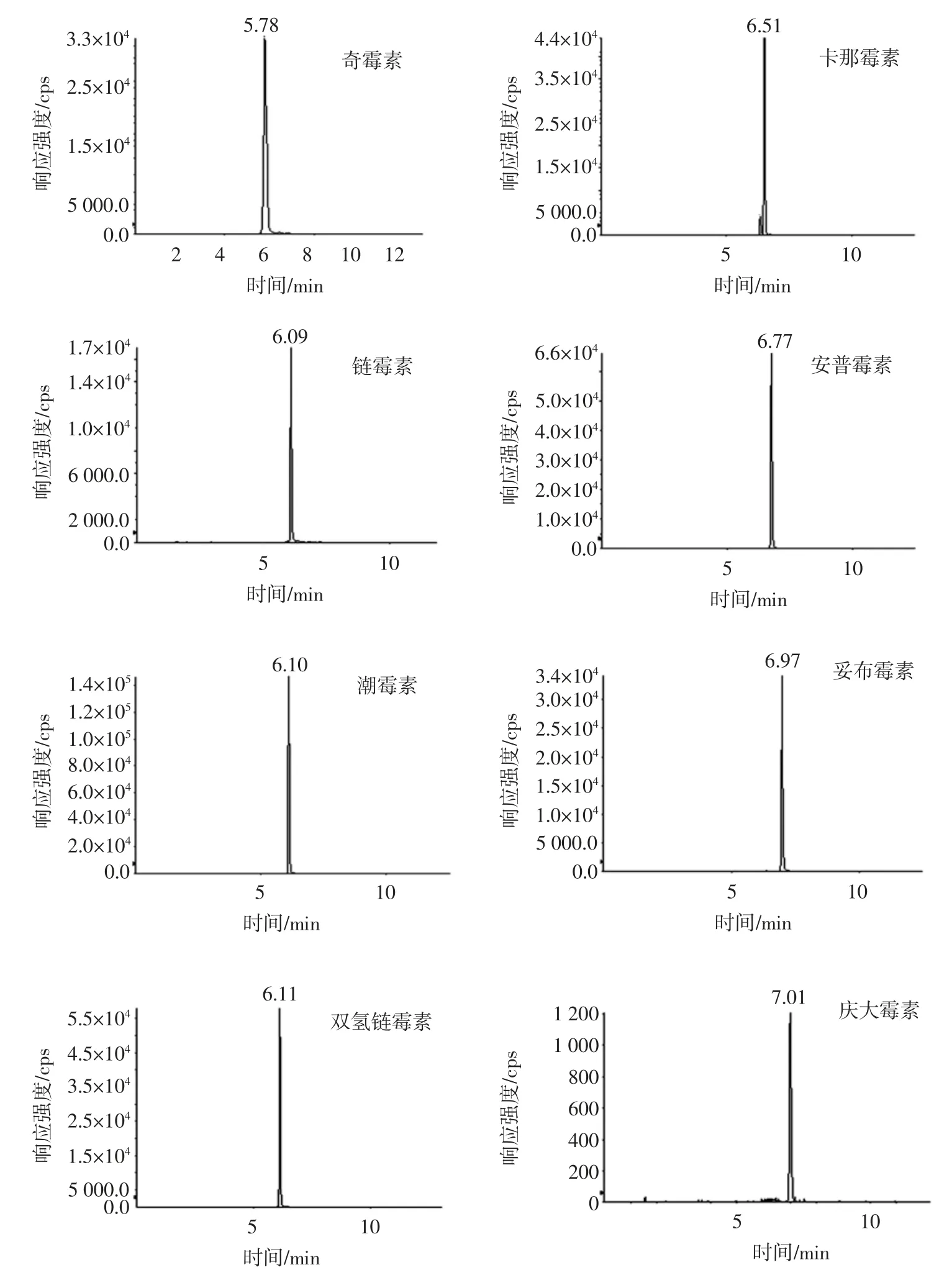
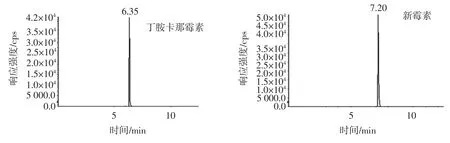
圖3 10 種氨基糖苷類藥物經親水作用色譜柱分離的提取離子色譜圖Fig.3 MRM chromatograms of ten aminoglycosides separated by HILIC column
2.6 回收率和精密度
在空白的豬肉、牛肉、豬腎和牛肝樣品中添加20、40、200 μg/kg 的丁胺卡那霉素、奇霉素、鏈霉素、雙氫鏈霉素、卡那霉素、慶大霉素和妥布霉素和100、200、1 000 μg/kg 的新霉素、潮霉素B 和安普霉素,以各自的定量離子對進行定量。
豬肉、牛肉、豬腎和牛肝中10 種氨基糖苷類藥物的添加回收率和精密度的結果見表3。
表3 的結果顯示,在豬肉、牛肉、豬腎和牛肝中分別添加1 倍、2 倍和10 倍定量限的10 種氨基糖苷類藥物的檢測回收率為73.9%~103.6%,相對標準偏差為3.3%~14.8%,可以滿足分析檢測的需要。
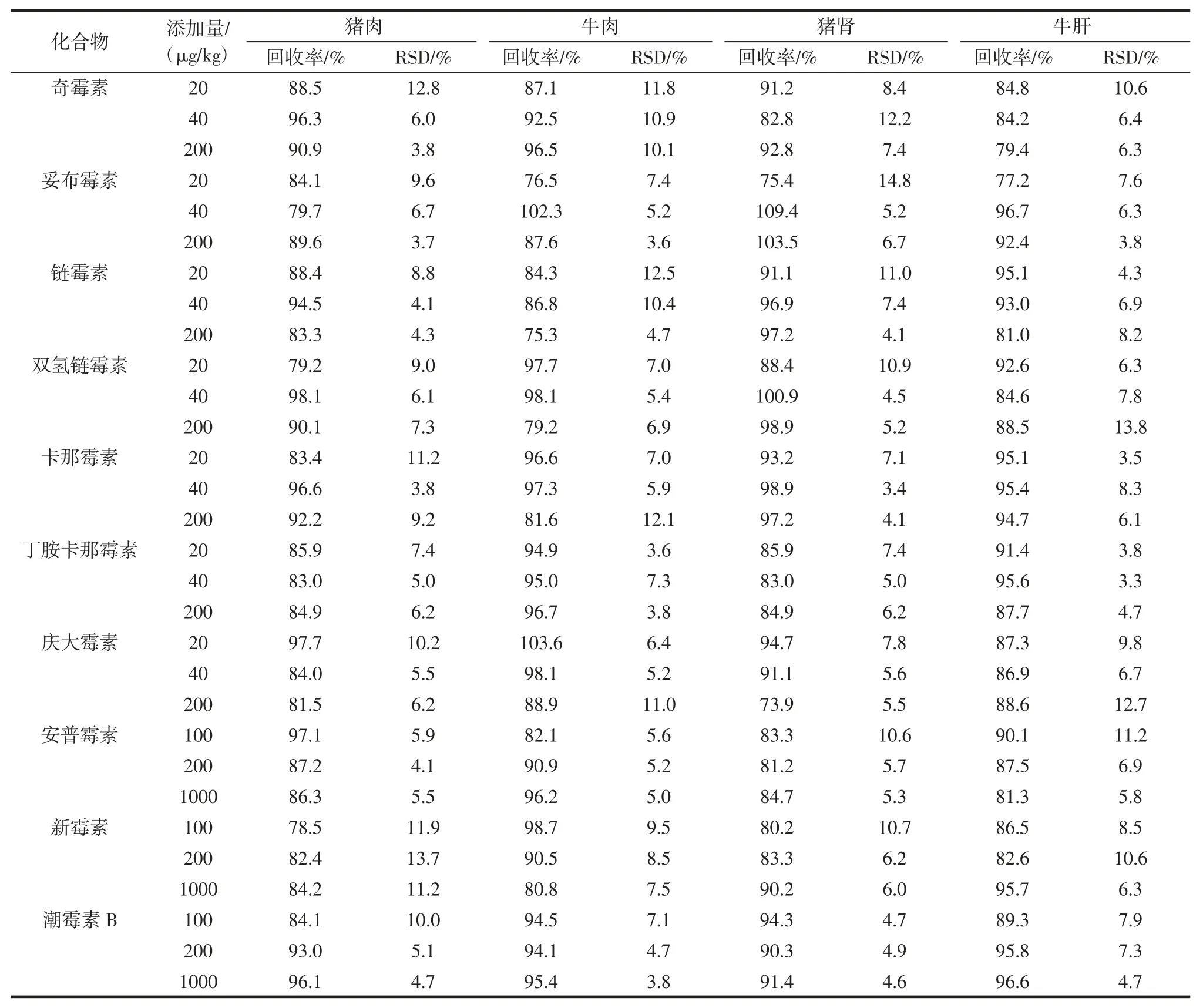
表3 不同基質中10 種氨基糖苷類藥物的添加回收和精密度(n=6)Table 3 Recoveries and RSDs of ten aminoglycosides spiked in different matrices(n=6)
3 結論
本研究針對質譜分析氨基糖苷類藥物時的常規反相色譜無法有效分析目標物的缺陷,通過4 種HILIC 機理色譜柱的比較和流動相及凈化定容溶液的優化,建立了動物源食品中10 種氨基糖苷類藥物殘留量檢測的親水作用色譜-串聯質譜方法。雖然親水作用色譜體系中的相互作用機制較為復雜,需要針對目標物進行流動相和預處理方法的反復調整,但與常規的反相色譜體系相比,本方法避免在流動相中使用大量離子對試劑,利于質譜檢測。本方法的靈敏度高,重現性好,適于推廣。
