胃腸道微生物種群與人類消化系統疾病相關性研究進展
2019-08-21 08:31:48劉德華孫寶林
生物學雜志 2019年4期
劉德華, 孫寶林
(中國科學技術大學 生命科學學院, 合肥 230026)
胃腸(GI)道具有250~400 m2,是人體和環境因子及抗原的最大接觸面之一[1]。在人的一生中,除了大約有60 t食物外,還有大量來自環境的微生物通過胃腸道,這些微生物對胃腸道的完整性產生了重要影響。細菌、古生菌和真菌在消化道的集落統稱為腸道微生物群,它們與宿主共同進化了數千年,形成了復雜而互利的關系。據估計,人體消化道承載的微生物數量高達1014個,近乎人類細胞的10倍[2]。通過16S rRNA基因測序方法檢測腸道微生物組,發現腸道微生物基因數達500萬,是人類基因組的150多倍[2]。因此,通常將宿主及寄生于其上的大量微生物稱為超級有機體[3]。
胃腸道作為具備代謝、免疫和內分泌功能的器官,可以與機體的其他器官相互作用、相互影響。微生物群可以增強腸道完整性、塑造小腸上皮、獲取能量、抵御病原體和調節宿主免疫等。然而,這些功能可能由于微生物組成的改變而被破壞,這種腸道微生物群的改變被稱為微生態失調。隨著分析這個復雜生態系統的方法越來越成熟,微生物群對胃及腸道疾病的影響及作用也越來越明確。腸道微生物與消化系統疾病具有一定相關性,通過對其組成、功能以及致病機制的研究,希望有助于疾病的防治和新治療方法的開發。本文概述了我們目前對人體胃腸道微生物群的發展和組成的認識,以及它與人體消化系統疾病的關系。
1 胃腸道微生物的研究方法
10年前,人們對胃腸道微生物群的了解大都來源于實驗室大規模培養。現在,隨著非培養、高通量以及低成本的測序方法的出現,對胃腸道微生物群檢測能力有了很大提高。目前針對細菌16S rRNA基因的靶向性測序是檢測腸道菌群的流行方法,16S rRNA基因存在于所有的細菌和古生菌中,并且包含9個高度可變的區域(V1-V9),可以依此對物種進行區分。近年來,16S rRNA基因測序已可以深入地分析該基因的短亞區,但是使用較短的讀取長度(100~250 bp)可能會引入錯誤[4]。而宏基因組測序以環境樣品中的微生物群體基因組為研究對象進行測序分析,具有較高的分辨率和靈敏度,可以更可靠地檢測分析微生物群的組成和多樣性。宏轉錄組可以直接獲得環境中微生物轉錄組信息,對微生物群的差異表達基因和差異功能進行分析[5]。利用這些新測序技術生成的大數據和先進的計算策略,如基因組裝配與基因發現軟件、統計建模與仿真、基因注釋工具等,可以對微生物群進行準確分析(圖1)。計算機和測序技術的蓬勃發展,極大地促進了整個人類微生物學領域的發展。
綜合MetaHit和人類微生物組計劃的數據可以獲得迄今為止人體相關微生物最全面的信息。這些研究匯總數據表明,從人體中共分離出2172種微生物,分為12個不同的門,其中93.5%屬于Proteobacteria、Firmicutes、Actinomycetes和Bacteroidetes。在人體中,有386種已鑒定的菌種為嚴格厭氧,通常定殖在口腔和胃腸道等黏膜區域[6]。目前已知Actinomycetes、Firmicutes、Facteroidetes和Proteobacteria在人體胃中占主導地位,而Proteobacteria、Firmicutes常見于腸道。共生腸道菌群具有多樣性、穩定性、抗性和恢復力強等特點,而非共生腸道菌群相對豐度較低,并缺乏共生性和多樣性[7]。
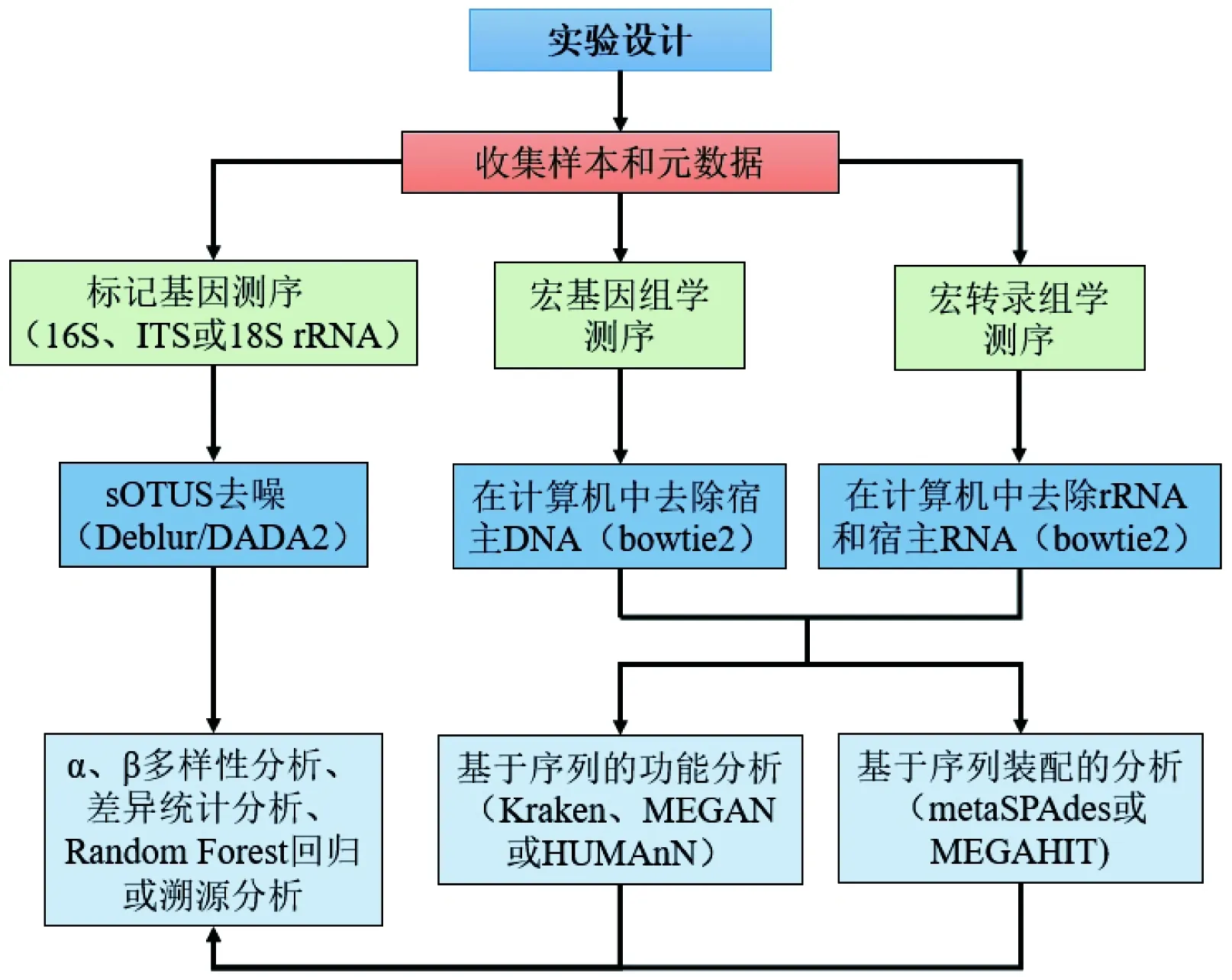
圖1 16S rRNA、宏基因組學和宏轉錄組學測序分析流程
胃腸道微生物群的多樣性并不像身體其他部位,如皮膚和口腔那樣豐富,并且胃腸道菌群顯示出高度的功能冗余[8]。有研究通過249個新測序和1018個已發表樣本組合鑒定出近千萬個基因,從而獲得了人類腸道微生物組的功能目錄。這項研究分析了不同國家人群腸道的菌群特征,證明腸道微生物群的組成由環境因素和宿主基因共同決定[9]。但不同組成的微生物群可能具有某種程度的功能冗余,以及相似的蛋白質或代謝產物譜。這一信息對于制定有關改善和塑造人類疾病中胃腸道微生物群的治療策略至關重要。
2 胃部微生物與胃部疾病
人體胃部微生物群鑒定和分類的研究最初是基于經典的微生物體外培養技術和胃標本,包括黏膜和胃液。在健康狀況下,人體胃部可以分離培養的菌屬主要有Clostridium、Lactobacillus和Veillonella。隨著高通量DNA焦磷酸測序、宏基因組測序和16S rRNA基因測序等先進的分子技術的出現,人們對于胃微生物組的生物多樣性有了更充分的了解[10]。基于這些測序分析發現人體胃部在健康條件下的菌屬主要有Prevotella、Streptococcus、Veillonella、Rothia、Pasteurellaceae、Fusobacterium、Actinomyces、Neisseria、Haemophilus和Porphyromonas,而且胃腔及胃體各部分的微生物群沒有實質性的差異[11]。
雖然胃部微生物群結構組成的形成機制尚不明確,但普遍認為飲食、抗生素、益生菌、長期使用質子泵抑制劑(PPIs)或H2拮抗劑和H.pylori感染等多種因素都會改變胃部微環境[12](圖2)。其中,長期使用PPI或H2拮抗劑會抑制胃酸分泌,對胃微生物群具有嚴重影響。早期研究報道,個體抗酸處理后胃內細菌會過度生長[13]。人體在補充維生素D3后胃部菌群多樣性增加,但腸道微生物群不會受到影響,說明某些因素可能會優先影響胃部微生物群的組成,而不會干擾人體其他部位的微生態[14]。研究表明遺傳背景似乎不會影響胃部微生物群的組成,同卵雙胞胎之間的胃部微生物群組成與不相關的個體相比沒有顯著區別[15]。另一個可能調節胃部微生態的因素是宿主的免疫狀態,因為微生物群的組成對人體免疫細胞有著重要影響,反之,宿主良好的免疫反應有助于調控胃部微生物群失調[16]。免疫抑制狀態、抗生素治療和胃液pH值大于4均可能引起胃部微生物群多樣性的降低,其中免疫抑制與Lactobacillus過度生長和Prevotella、Fusobacterium豐度降低有關[17]。
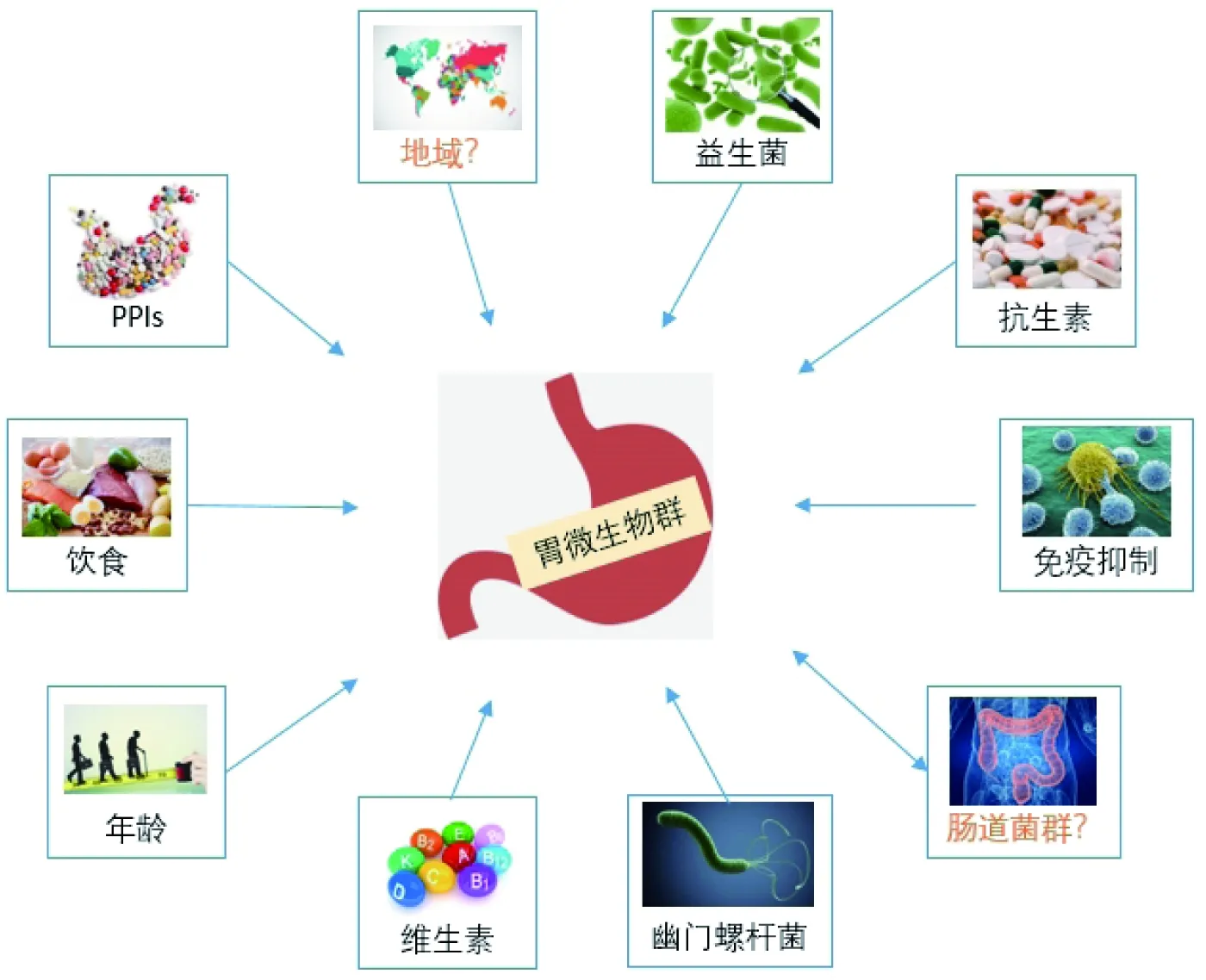
圖2 胃微生物群的影響因素
幽門螺桿菌(Helicobacterpylori)是一種常見于人類胃部的細菌,是消化性潰瘍、胃癌和黏膜相關淋巴組織(MALT)淋巴瘤的主要致病菌。胃癌是全球第四大常見癌癥,其大多數病例發生在發展中國家。H.pylori感染是胃癌發生發展的最大危險因素,占全球胃癌的75%[18]。H.pylori具有多種毒力因子,其中細胞毒素相關基因A(CagA)、空泡細胞毒素(VacA)和外膜蛋白(OMPs)直接或間接與H.pylori的致癌性有關[19]。除了這些毒力因子外,胃部感染H.pylori所產生的慢性炎癥也是促進胃癌發生發展的關鍵因素[20]。
H.pylori和胃微生物群之間的相互作用還沒有完全闡明,但H.pylori的定殖可能誘發特定胃微生物群組成的變化,引起胃微生物群的失調。H.pylori感染陽性患者的胃微生物組與未感染個體的胃部微生物組有所不同,其胃部non-Helicobacter、Proteobacteria和Spirochetes豐度較高[21]。在有消化不良癥狀的兒童中,H.pylorie感染陰性患者的菌群多樣性更高,主要類群有γ-Proteobacteria、β-Proteobacteria、Bacteroidia和Clostridia[22]。但目前還不清楚是H.pylori感染本身促進有害微生物的生長,還是微生物群失調引起黏膜或胃腔的變化,從而為H.pylori的定殖創造了有利條件,很有可能存在一種雙向的相互作用。
H.pylori在胃癌發生中的作用毋庸置疑,然而有證據表明胃部微生物群中的其他微生物也可能與胃上皮細胞的癌變有關。胃癌組織具有獨特的微生物譜,與正常胃組織相比胃癌組織的微生物群多樣性有所降低,而根除H.pylori后菌群多樣性可以恢復[23]。胃部Lactobacilluscoleohominis和Lachnospiraceae的增加,Porphyromonas、Neisseria和Streptococcussinensis的減少均可能與胃癌的發生具有相關性[24]。此外,一項對胃癌患者胃大部切除術前后胃微生物群的調查研究顯示,胃癌患者胃部微生物群的多樣性在手術后具有特異性的改變,表現為Proteobacteria和Actinobacteria數量減少以及Firmicutes和Bacteroidetes數量增加[25]。最近有研究發現在胃癌發生發展的不同階段(從淺表性胃炎、萎縮性胃炎、腸上皮化生到胃癌),胃部微生物群出現顯著失調,并且口腔微生物Peptostreptococcusstomatis、Streptococcusanginosus、Parvimonasmicra、Slackiaexigua和Dialisterpneumosintes相互影響逐漸形成致癌作用越來越強的特異性微生態,體現了口腔致病性菌群在胃癌的無創診斷中的潛在應用價值[26]。
血清胃蛋白酶原I與胃蛋白酶原II比值降低與上消化道微生物群多樣性的降低有關,這一比值也與胃癌的發生具有相關性[27]。此外,胃癌患者的胃液中亞硝酸鹽的含量高于健康對照組,可能是其胃內較多的厭氧菌促進了胃內亞硝酸鹽的積累[28]。新的研究發現,與慢性胃炎相比,胃癌微生物群的物種多樣性降低、H.pylori數量減少以及具有潛在遺傳毒性的微生物比例升高,同時由PICRUSt功能預測得知胃癌微生物群硝酸鹽還原酶和亞硝酸鹽還原酶的功能增強[29]。
雖然我們對胃微生物群的認識有了很大的拓展,但目前對胃微生物群的研究大多局限于小群體中的個體,因此需要設計包括大量個體的縱向前瞻性研究,明確闡明胃微生物群在疾病發展中的作用。此外,還需要進一步研究抗炎藥物、類固醇和免疫抑制劑在H.pylori存在或不存在的情況下對胃微生物群的影響。同時需要開發新方法,能夠快速、準確識別并描述胃微生物群的形成與功能,例如,能夠測量微生物代謝活性的方法將有助于開發適用于臨床環境的微生物群檢測技術。而了解胃癌發生發展過程中微生物群的動態,將為潛在的預防、診斷和治療胃癌的臨床應用開辟新的方向。
3 腸道微生物與腸道疾病
近十年來,微生物學研究的熱潮為人們描繪出了腸道微生物的組成和某些功能的藍圖。人們在研究炎癥性腸病(IBD)和結直腸癌(CRC)的腸道微生物群組成方面做出了巨大努力和重大進展,但是由于人類腸道微生物群的復雜性,許多微生物功能方面的問題仍未解決。
炎性腸病(IBD),包括克羅恩病和潰瘍性結腸炎,在全世界范圍內發病率逐漸增加[30]。IBD的特征是免疫介導的腸道慢性炎癥,受遺傳易感性和環境因素如飲食、抗生素的使用等影響[31]。臨床發現抗生素可以治療IBD,這與腸道菌群可能引起炎癥反應的觀點相一致[32]。有研究發現,克羅恩病患者的腸道微生態失調反映了炎癥的存在及其嚴重程度,同時微生態失調與飲食和抗生素使用等因素有關聯[33-34]。因此,腸道微生物群改變可能在IBD的早期出現并導致疾病的發生,而炎癥在內的環境因素可能通過改變腸道的代謝條件進一步導致微生態失調。IBD患者微生物群會發生整體變化,但是某些特定病原體,如Enterobacteriaceae以及Mycobacteriumavium的亞種paratuberculosis可能是IBD的潛在病原體[35]。并且在潰瘍性結腸炎患者腸道中可以分離出高侵襲致病性菌株Fusobacteriumnucleatum[36]。腸道微生物群可能誘導IBD發生,但目前為止的研究都只闡述了相關性而未證明因果關系。IBD中的微生態失調在很大程度上反映了復雜微生物群對腸道炎癥等環境壓力的響應,同時與炎癥和病原體定殖相關的微生物代謝改變可能促進微生態失調。
腸道微生物群組成的變化導致的代謝產物改變,可能在IBD發病過程中起作用。代謝組學研究表明,健康個體和IBD患者的腸道微生物群之間有12%的代謝途徑明顯不同。具體而言,IBD患者微生物群中氨基酸合成和碳水化合物代謝途徑減少,營養攝取和毒力的分泌途徑增加[37]。生物信息學分析顯示,與健康個體相比,IBD患者腸道微生物群中膽鹽水解酶(BSH)的分泌顯著降低, Firmicutes占比顯著下降[38]。與IBD相關的另一種細菌代謝途徑是某些碳水化合物經微生物發酵產生的短鏈脂肪酸(SCFAs)。Clostridia產生的SCFAs可以激活G蛋白偶聯受體以及通過表觀遺傳效應增強腸黏膜中調節性T(Treg)細胞功能,從而促進免疫耐受的恢復并減少結腸炎的發生[39]。
糞菌移植(FMT)成功治療艱難梭菌感染激起了人們使用FMT治療IBD的研究興趣[40]。然而,目前為止FMT治療IBD的臨床結果不一致,可能是因為與艱難梭菌感染引起的結腸炎相比IBD的復雜性更高[41-43]。在使用安慰劑作為對照的兩項治療潰瘍性結腸炎的隨機試驗表明,在緩解潰瘍性結腸炎成年患者的臨床癥狀方面,FMT明顯比安慰劑更有效[44-45]。但是,在治療腸應激綜合征(IBS)的隨機雙盲臨床試驗中,FMT沒有表現出很好的療效,同時FMT組出現了不良反應[46]。因此,FMT(短期和長期)的安全性和持久性、對免疫抑制患者的影響、最有效的管理方式以及如何選擇合適的供體和受試者等方面仍然存在疑問。為了更好地確定FMT在IBD治療中的作用,必須進行更大規模臨床隨機對照試驗。
結腸直腸癌(CRC)常與腫瘤及其鄰近黏膜處的微生物群失調有關,但CRC進展中的優勢菌種仍不清楚。研究分析發現CRC患者糞便中菌群多樣性降低,其中纖維發酵相關菌類Clostridia減少,口腔共生Fusobacteriumnucleatum和Porphyromonas增加[47]。有研究表明F.nucleatum通過干擾TLR4和MyD88信號通路使CRC患者產生對抗腫瘤藥物Oxaliplatin和5-FU的耐藥性并更容易復發,F.nucleatum還可以靶向microRNA激活自噬途徑從而改變化療反應[48]。F.nucleatum感染CRC細胞可增加其增殖率和侵襲活性,同時F.nucleatum還具有誘導小鼠產生異種移植瘤的潛能[49]。某些Escherichiacoli可以促進腸道炎癥的發生,產生具有致癌性的毒素如大腸桿菌素[50]。而黏膜相關E.coli在CRC組織中更為普遍,可能與腫瘤分期和預后有關[51]。宏基因組研究發現E.coli和F.nucleatum在CRC發生發展中的作用不盡相同[52]。例如,臨床CRC患者的F.nucleatum相關分離物未能誘發小鼠炎癥和癌癥,而產大腸桿菌素E.coli卻能促進ApcMin/+;Il10-/-小鼠腫瘤發生,但該臨床表現可能受F.nucleatum定殖程度和腸道腫瘤位置影響[53]。Bacteroidesfragilis構成人體共生微生物群的1%~2%,其衍生毒素(BFT)可以引起炎癥性腹瀉和炎癥相關腫瘤的發生[54]。分泌BFT的腸毒性B.fragilis(ETBF)可由炎癥性Th17細胞驅動,誘導ApcMin/+小鼠產生結腸炎和結腸腫瘤[55]。使用頭孢西丁治療可完全清除ETBF的定殖,從而減少小鼠IL-17A表達和結腸腫瘤發生[56]。BFT還可能上調精胺氧化酶產生活性氧并引起DNA損傷,進而導致炎癥和腫瘤發生[57]。多項動物實驗證明F.nucleatum、產大腸桿菌素E.coli和腸毒性B.fragilis及其代謝產物與CRC的發生發展具有相關性,但其因果關系還需進一步的臨床研究。
研究發現Parvimonasmicra和Solobacteriummoreii等也與CRC發生相關,使用腸道微生物群中20個菌種的基因和兩種酶基因(F.nucleatum的丁酰輔酶A脫氫酶和P.micra的RNA聚合酶亞基β)可以顯著地區分出CRC患者[58]。而使用F.nucleatum、Bacteroidesclarus、Roseburiaintestinalis和Clostridiumhathewayi構建模型預測受試人群中CRC患者,其敏感性>90%,特異性>80%[59]。結腸腫瘤與癌旁黏膜間的菌群失調比例相似,某些特征菌群組成甚至存在重疊,說明結腸在癌癥環境中已經出現相當程度的微生物群失調,因此CRC的糞便評估能充分反映黏膜活檢的微生物組成[60]。上述研究表明,通過檢測人體糞便中某些菌株及其衍生因子可作為CRC預防及診斷的篩選工具。
腸道微生物多樣性降低是許多腸道和腸外疾病的特征,共生微生物群中某一類細菌的增加或減少以及特定菌種豐度改變都可能促進炎癥以及腫瘤的發生,但其相關機制仍需進一步研究。值得注意的是,IBD患者的腸道微生物群表現出與CRC患者相似的組成改變,看來不僅是炎癥,菌群失調也可能是CRC和IBD發生的關聯因素之一[61]。因此,深入了解宿主微生物群的互利共生性和環境信號引起的干擾,對開發腸道疾病的新型治療方法具有重要意義。
4 總結
對人體胃腸道微生物群的認識在快速發展,目前宏基因組關聯分析可以確定微生物群組成的主要影響因素,并將單個微生物在物種水平上與宿主疾病、生活方式和生理學聯系起來。而宏轉錄組學方法的應用也極大地促進了我們對胃腸道微生物群組成及其代謝產物影響人類健康和疾病相關機制的理解。胃腸道微生物群與宿主之間存在著密切的共生關系,消化系統的眾多疾病環境中,均可以觀察到其微生物群組成的紊亂。無論胃腸道微生物群失調是疾病的起因還是結果,都可能加劇疾病的進展,并影響相關的治療策略。
隨著對胃腸道微生物群組成功能認識的不斷深入,通過調控微生物群組成來改善患者疾病狀況的治療方法受到廣泛關注。然而目前對于健康微生物組仍未達成共識,有效、精準的微生物治療手段還處于起步階段,還需要推動更多更大規模的縱向和介入性研究,使用更新的方法學,包括多種基因組技術和新的體外方法,同時還要更多地關注微生物組本身的相互作用。這些研究將進一步加深對胃腸道微生物組的理解,闡明其致病機制,開發更安全有效、有針對性的腸道菌群療法,從而更好地預防和治療疾病。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
汽車工程學報(2017年2期)2017-07-05 08:13:02
中國組織化學與細胞化學雜志(2016年3期)2016-02-27 11:15:35
中國衛生標準管理(2015年3期)2016-01-14 03:41:46
醫學研究雜志(2015年6期)2015-07-01 17:40:49
醫學研究雜志(2015年9期)2015-07-01 17:28:27
