以質粒為基礎的同源重組技術在葡萄球菌基因敲除中的應用
2019-08-07 05:52:02武有聰孟媛媛丁百興2韓海燕2滌2
中國人獸共患病學報 2019年7期
武有聰,2,孟媛媛,丁百興2,韓海燕2,瞿 滌2,白 麗
研究細菌基因功能的方法主要有基因敲除(基因組中直接去除某基因,位點特異)、反義RNA干擾(降低mRNA表達量,影響因素多)、轉座子插入突變(插入位點隨機,篩選繁瑣,存在多位點插入突變)、基因過表達等技術,但最可靠而常用的方法仍是基因敲除[1-2],即選擇合適的質粒,將靶基因兩側的同源序列克隆至載體上,轉入宿主菌后通過同源重組實現對靶基因的置換敲除。由于微生物的多樣性,將外源DNA導入宿主菌并完成同源重組的策略也千差萬別。目前,未見有適合于多種宿主菌基因敲除的通用重組系統的報道。因此,需要根據宿主菌的特點來構建相對特異的基因敲除系統,用于基因功能研究。
葡萄球菌屬(Staphylococcusspp.)是引起醫院內感染的重要病原菌[3-5],由于其GC含量較低(mol%約28%~35%),通過同源重組進行基因敲除相對較困難,常常受限于轉化效率低、重組頻率低、突變株篩選繁瑣等困難,是葡萄球菌致病基因功能研究的難點。隨著分子生物學技術的發展,用于葡萄球菌基因敲除的重組系統也得到不斷改進,基因敲除的過程也變得簡捷。作者結合本實驗工作,對葡萄球菌基因敲除中采用的以質粒為基礎的同源重組技術進行如下綜述。
1 同源重組技術敲除細菌靶基因的機制
將目的基因(待敲除的靶基因)兩側的同源序列(分別稱為同源臂A/B)克隆至復制起始子缺陷的質粒中,一般在兩段同源臂之間含有一段抗性基因(圖1A)。在非容納條件下,質粒通過同源重組整合至細菌基因組中(圖1B)。在容納條件下,通過質粒的滾環復制發生單交換和雙交換,單交換重組時形成含有野生型和突變型同源序列的部分二倍體細胞,靶基因敲除不成功。雙交換重組時,隨著質粒的切除,要么突變型同源序列留在細菌基因組中,帶有野生型的質粒被切除,靶基因敲除成功(圖1C-1);或者野生型序列留在細菌基因組中,同源重組質粒切除,靶基因敲除不成功(圖1C-2)。在大多數菌株中,同源重組是一個低概率事件,所以需要插入抗性標記幫助篩選突變株。除非是重組質粒攜帶反向選擇標記,否則質粒切除或丟失發生的概率都非常低,需要大量精力來篩選突變株[6-7]。
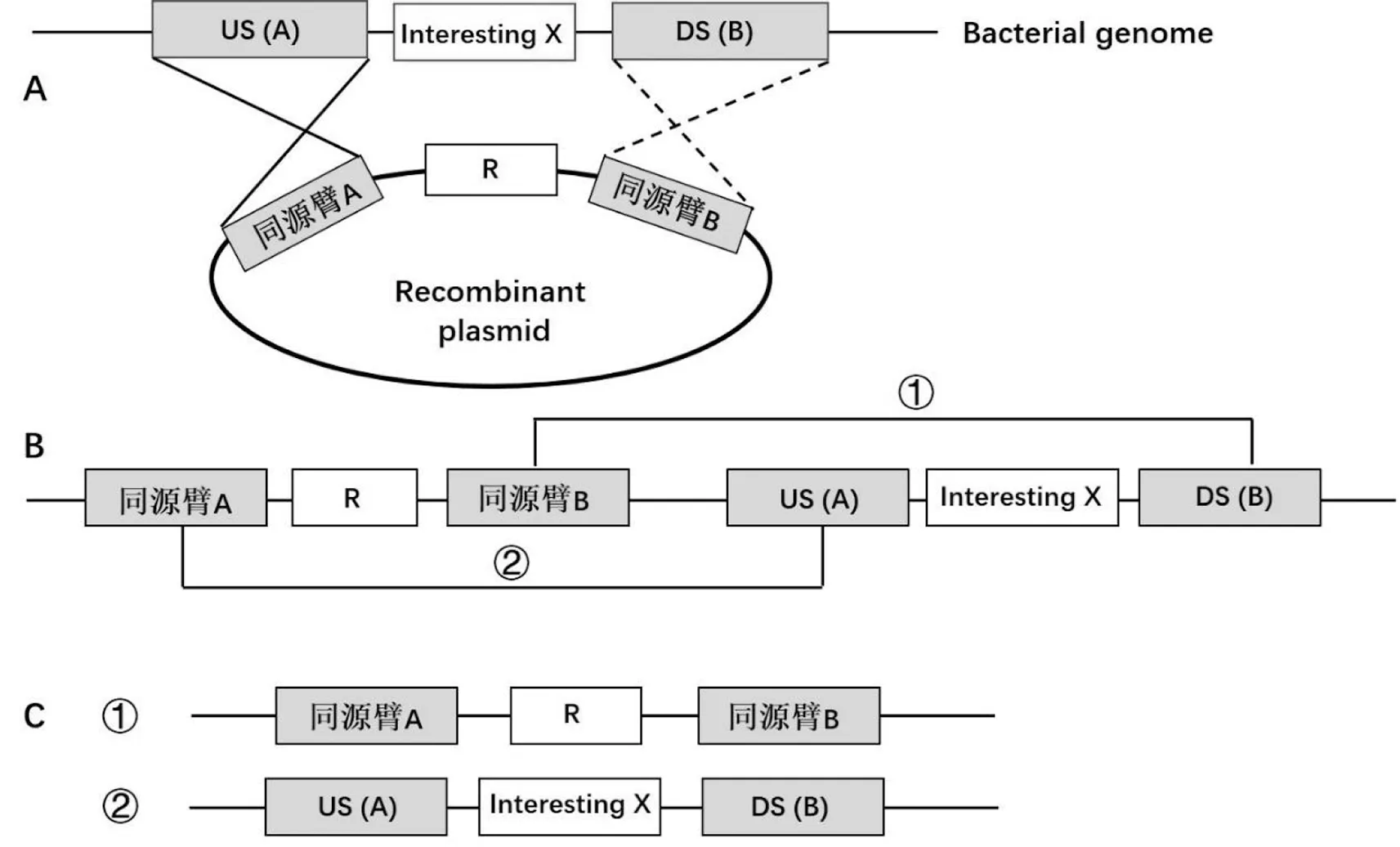
A為同源重組質粒的構建;US為上游同源臂; DS為下游同源臂; R為耐藥基因. B為重組質粒DNA與細菌基因組整合;C為雙交換重組時質粒DNA的切除。圖1 同源重組在細菌基因敲除中的作用Fig.1 Role of allelic replacement in the gene deletion
2 pBT-2重組質粒在葡萄球菌基因敲除中的應用
2.1pBT-2重組質粒構建及其特點 由于質粒DNA與宿主菌基因組的整合是隨機的,發生整合的效率可高可低,所以對克隆載體的選擇非常重要。Reinhold等[8]利用溫度敏感型質粒pTV1Ts(40 ℃質粒消除成功率高,pE194質粒來源)構建同源重組質粒,用于肉葡萄球菌(S.carnosus)和木糖葡萄球菌(S.xylosus)基因敲除的研究。將pTV1Ts 質粒來源的BamHI-PstI酶切片段(4.0 kb)與pBR322質粒來源于的BamHI-PvuII酶切片段(2.7 kb)連接形成pBT1質粒,前一片段含有質粒pE194的復制子和pC194的氯霉素乙酰基轉移酶基因(cat194),后一片段含有內酰胺酶基因和質粒ColE1復制起始子。pBT1質粒獲得大腸桿菌和葡萄球菌的復制能力,在大腸桿菌內表現為氨芐青霉素抗性(cat194表達相對較低,不推薦使用氯霉素抗性標記),葡萄球菌內氯霉素抗性(在亞抑菌濃度作用下葡萄球菌轉化子的涂板陽性率可提高約3倍)。再將pUC18來源的HindIII-BglI片段連接入pBT1,得到含有多克隆位點(12個酶切位點)的pBT2質粒,多克隆位點位于轉錄終止子T1附近。pBT2質粒在大腸桿菌和葡萄球菌中結構較穩定,30 ℃條件下能穩定存在,40 ℃以上容易丟失,比較適合于葡萄球菌靶基因的突變研究。
2.2pBT2重組質粒在葡萄球菌基因敲除中的應用 Reinhold等[8]用pBT2質粒敲除肉葡萄球菌磷酸轉移酶系統(phosphotransferase system, PTS)基因ptsI,為方便突變株的篩選,作者在同源臂構建過程中引入了紅霉素抗性基因(ermB)。PCR分別擴增ptsI基因兩側大約1 kb片段(上/下游同源臂),與pEC7質粒來源的EcoRI-PstI酶切片段(含ermB)克隆連接至pBT2質粒,形成的重組質粒pBTIE1轉入肉葡萄球菌,30 ℃條件下,氯霉素(Cm 20 g/mL)敏感,紅霉素(Erm 10 g/mL)耐藥,并且不能分解甘露醇和果糖的菌株為疑似突變株。經過Southern Blot驗證ptsI基因被敲除,并且證實基因組上沒有質粒的序列。獲得突變株菌落的比例約8%~12%(個體試驗差別大)。另外,又用pBT2質粒敲除木糖葡萄球菌的蔗糖酶基因scrB,不同的是上/下游同源臂大小分別為1.7 kb和2 kb,基因敲除效率42%~57%。Egeter等[9]用pBT2質粒敲除葡萄球菌的其它基因,發現敲除效率從1%~50%不等。
pBT2不僅可以用于肉葡萄球菌和木糖葡萄球菌的基因敲除,也可以用于金黃色葡萄球菌和表皮葡萄球菌。本實驗室[10-11]用pBT2成功敲除表皮葡萄球菌雙組分系統lytSR和arlRS。PCR擴增lytSR和(或)arlRS上/下游同源臂(lytSR同源臂約1.8 kb,arlRS同源臂約0.9 kb~1.2 kb),克隆形成pBT2-△lytSR/△arlRS重組質粒,重組質粒經金黃色葡萄球菌RN4220限制性修飾后轉入表皮葡萄球菌SE1457。30 ℃培養(BM培養基,Erm 10~20 g/mL)至穩定期末,1∶100轉接(BM,Erm 2.5 g/mL),42 ℃培養過夜,再經多次轉接后(BM,無抗生素)取適量涂板,紅霉素耐藥(2.5 g/mL)和氯霉素敏感(10 g/mL)的克隆為疑似突變株。疑似突變株經PCR、RT-PCR及測序鑒定敲除成功。該方法篩選工作繁瑣,敲除效率比上述文獻報道的明顯偏低,往往需要幾個月的篩選才能得到突變株。
3 pMAD重組質粒在葡萄球菌基因敲除中的應用
3.1pMAD重組質粒的構建及其特點 一般敲除突變株的篩選是通過同源重組將質粒上的抗性基因與宿主菌基因組中的靶基因進行置換,使宿主菌在缺失靶基因的同時獲得抗性基因,通過抗性標記直接篩選疑似突變株,這種方法稱為正向選擇或陽性選擇(positive selection)[12]。但由于同源重組的概率非常低,發生單交換的質粒從基因組中切除較困難,給篩選工作帶來巨大的麻煩。Tamura等[13-14]首先使用反向選擇(counter-selection)來篩選疑似突變株,如將與蔗糖(sacB)或谷氨酰胺代謝相關的基因(glnQ)克隆至重組質粒中,重組質粒切除不成功的菌株在適當底物作用下高表達sacB或glnQ基因,導致毒性代謝產物堆積而生長抑制,平板上不能形成克隆,重組質粒與菌株基因組進行二次交換,成功敲除靶基因的菌株生長不受影響而更容易被篩選出來。但由于葡萄球菌基因組中含有與蔗糖和谷氨酰胺代謝相關的基因,不能通過基因sacB或glnQ的表達來反向選擇。
Arnaud等[15]成功構建了可通過反向選擇來敲除葡萄球菌靶基因的重組質粒pMAD。首先,用限制性內切酶ClaI將質粒pE194ts(pE194來源,溫度敏感型復制起始子)線性化,克隆至質粒pBR322中,形成穿梭質粒pE194ts:pBR322。在NruI和EcoRV之間插入3個PCR片段:1)從質粒pMTL22來源的多克隆位點,NaeI和BglII的酶切片段;2)以金黃色葡萄球菌(S.aureusRN6390)基因組為模板,PCR擴增clpB基因(編碼Clp-Hsp100 ATP酶)的啟動子區,并加上BglII和BamHI酶切位點;3)嗜熱脂肪芽胞桿菌(Bacillusstearothermophilus)不含啟動子區的bgaB基因(編碼耐熱半乳糖苷酶),BamHI和StuI的酶切片段。上述3個片段連接入質粒pE194ts:pBR322,獲得重組質粒pMAD(G-菌中氨芐青霉素抗性,G+菌中紅霉素抗性)。pMAD質粒帶有pE194ts復制起始子(溫度敏感性)和bgaB基因,通過clpP啟動子(pclpB),編碼耐熱的半乳糖苷酶,在含X-gal平板上形成藍色菌落。這技術本身并不能影響同源重組的頻率,但對于質粒丟失或從基因組中切除菌株的篩選非常有利。
3.2pMAD重組質粒在葡萄球菌基因敲除中的應用 Arnaud等[15]利用pMAD敲除金黃色葡萄球菌vraFG基因。PCR擴增vraFG上/下游同源臂(約0.6 kb和0.9 kb),壯觀霉素耐藥基因spc(1.3 kb,pIC333質粒來源),重組PCR將3個PCR片段克隆至pMAD,構成重組質粒pMAD△FG,經鑒定后電擊轉入金黃色葡萄球菌(S.aureusMu3)。從TSA平板(含X-gal)上挑取1個藍色菌落轉接于無抗性TSB培養基,30 ℃震蕩培養2 h后變溫至42 ℃繼續培養6 h,取適量涂板于TSA(含X-gal和抗生素),42 ℃過夜培養。壯觀霉素(100 g/mL)耐藥、紅霉素(5 g/mL)敏感的白色菌落即為vraFG疑似突變株。據統計,1%~5%白色菌落為敲除突變株,還有大量的淡藍色菌落,可能由于單交換重組后質粒DNA與宿主菌基因組整合形成。作者還用pMAD敲除金黃色葡萄球菌sarA、sigB基因。
本實驗室[16-17]用pMAD質粒敲出表皮葡萄球菌SE1457雙組分系統saeRS。PCR擴增saeRS上/下游同源臂(約1.0 kb和1.3 kb),同源臂間連接壯觀霉素耐藥基因spc,酶切片段克隆至pMAD載體中,構建重組質粒pMAD△saeRS,轉入宿主菌SE1457中。挑取藍色單菌落于BM培養基(Spc 50 g/mL),43.5 ℃震蕩培養24 h,取適量菌液涂于BM平板(含X-gal及抗生素),43.5 ℃培養過夜,壯觀霉素耐藥,紅霉素敏感的無色菌落即為敲除突變株;涂板同時,轉種于新鮮BM培養基,相同條件下繼續震蕩培養,直至篩選出突變株為止。該技術通過藍白斑篩選,大大降低篩選的工作量,通常也需要1~2月可獲得突變株。
pMAD質粒還可用于一些G+菌的基因敲除,如單核細胞增多性李斯特菌(Listeriamonocytogenes)和蠟樣芽胞桿菌(Bacilluscereus)等[18-19],但由于pE194來源的復制起始子在鏈球菌、腸球菌、乳球菌中不工作,限制了該質粒的應用。
4 pKOR1重組質粒在葡萄球菌基因敲除中的應用
pMAD質粒的使用,通過顏色反應更容易實現同源重組突變株的篩選,但該質粒對重組概率及突變株篩選均無直接影響[15]。SecY是一種膜蛋白,屬于SecYEG轉位酶家族中的成分之一,能將含有前體蛋白的信號肽通過細胞膜轉移至細胞外,與細胞蛋白的分泌密切相關,SecY的表達是細菌生長所必需的[20-21]。因此,通過secY反義RNA的表達能夠抑制平板上菌落的形成。pKOR1重組質粒就是利用secY反義RNA的表達來實現對敲除突變株的反向選擇,大大地提高了同源重組的概率和突變株篩選的效率[21]。
4.1pKOR1重組質粒的構建及其特點 為構建secY反義RNA基因盒,通過PCR從金黃色葡萄球菌RN4220基因組中擴增含有核糖體結合位點的secY基因(570 bp),PCR產物經SmaI酶切后反向插入pYJ335質粒中Pxyl/tetO啟動子的下游,形成重組質粒p335Y-R,并將p335Y-R電擊轉入RN4220后,在脫水四環素(ATc)誘導作用下,平板上無克隆形成證實該質粒中secY反義RNA能正常工作[22]。
PCR擴增p335Y-R質粒中反義secY基因盒,酶切后連接入pTS1質粒,形成pTS1-HF質粒(含四環素抗性基因tetR,同時含有與復制基因rep轉錄相反的secY基因)。PCR擴增pDONR221質粒(Invitrogen)噬菌體基因盒,經T4 DNA聚合酶和多聚核苷酸激酶處理后插入pTS1-HF質粒(破壞了抗性標記cat基因),形成pHF-ccd質粒。PCR擴增pC194質粒中的cat基因,克隆至pHF-ccd質粒,形成重組質粒pKOR1。
pKOR1質粒具有噬菌體基因盒(Gateway technology, Invitrogen),不需要限制性內切酶和連接酶即能實現同源序列的快速克隆。重組盒子有2個關鍵成分(attP位點和ccdB位點)。在噬菌體整合酶和大腸桿菌整合宿主因子(E.coliintegration host factor, IHF)的作用下,attP序列能與DNA插入序列上的attB序列進行重組,形成新的重組質粒。ccdB基因編碼大腸桿菌解旋酶抑制劑,當DNA片段與pKOR1整合后,重組形成的pKOR1質粒丟失ccdB基因,重組子生長不受影響而大量增殖;整合不成功的pKOR1質粒,通過ccdB基因的表達生長受到抑制,簡化了重組質粒pKOR1的構建。
4.2pKOR1重組質粒在葡萄球菌基因敲除中的應用 Bae等[22]在不使用抗性標記的條件下,用pKOR1質粒對金黃色葡萄球菌(S.aureusNewman)rocA進行框內敲除(in-frame deletion,即基因敲除范圍限于開放讀碼框內)。PCR分別擴增(引物5′端加上attB序列)金黃色葡萄球菌rocA基因上/下游同源臂(約1.0 kb),同源臂片段間通過SacII連接后直接與pKOR1質粒進行重組(BP ClonaseTMenzyme mix, Invitrogen),獲得重組質粒pKOR1-△rocA,電擊轉化入金黃色葡萄球菌中,得到菌株NM(pKOR1-△rocA)。菌株NM(pKOR1-△rocA)過夜培養物1∶1 000接種于預熱TSB(Cm 10 g/mL),43 ℃(非容許溫度,質粒DNA整合至宿主菌基因組中)震蕩培養過夜,取適量劃線接種于TSA(預熱,Cm 7.5 g/mL),43 ℃培養過夜。挑取1個菌落接種于TSB(含Cm 10 g/mL),30 ℃培養過夜。取適量菌液稀釋(10-4)后分別涂于不同脫水四環素濃度TSA平板(ATc=0, 1, 2 g/mL),30 ℃培養2 d。在不含ATc的TSA平板上有大量的菌落形成,在ATc為2 g/mL TSA平板上無菌落形成(可能是葡萄球菌的最低抑菌濃度),1 g/mL TSA平板上約1%的細胞形成菌落,菌落大小不一,提示1 g/mL ATc是誘導反義secYRNA表達,選擇pKOR1質粒缺失菌株的最佳濃度。隨機挑選10個疑似突變株,經PCR驗證8株成功敲除rocA基因(獲得突變株效率80%)。作者比較了傳統pTS1質粒和pKOR1質粒在金黃色葡萄球菌prsA基因敲除中的效率,前者介導的同源重組中,累積7000個單交換整合的菌株中均未篩選出prsA突變株;后者介導同源重組概率約4%,進一步提示pKOR1是葡萄球菌同源重組敲除靶基因效率較高的質粒。
本實驗室[23-25]利用pKOR1成功敲除表皮葡萄球菌(SE1457)呼吸相關雙組分系統srrAB、萬古霉素耐藥相關雙組分系統vraSR、磷酸鹽代謝調節子phoU以及聚集生物膜形成調節子abfR基因等,敲除效率約30%~80%。此外,還用pKOR1成功敲除金黃色葡萄球菌臨床株(SA75)附屬基因調節子agr[26]。一般1~2周可獲得敲除突變株。
5 結 語
利用同源重組技術進行基因敲除是研究單基因功能的理想方法,該技術在G-桿菌中應用非常廣泛,如大腸桿菌中噬菌體Red同源重組系統,該系統所需同源臂小(40~60 bp),可以直接設計在引物上,重組效率高。葡萄球菌是常見的G+球菌代表,由于細胞結構及胞內酶系統的差異,導致基因敲除效率較G-桿菌困難。本室常采用以質粒為基礎的同源重組技術來敲除葡萄球菌的致病基因,不同實驗者或同一實驗者敲除不同的靶基因,敲除效率差別很大。據報道,宿主菌的生長時相,培養基的類型,篩選溫度及同源臂大小等因素均可影響敲除效率,但尚不清楚敲除效率與上述因素的直接量效關系。通常情況下,選擇TSB或BM培養基;常規細菌培養選擇37 ℃,質粒DNA整合入宿主基因組溫度為42 ℃~43 ℃,質粒切除或丟失溫度為30 ℃;同源臂大小約1 kb左右(0.3~2.0 kb)。早期使用pBT2質粒進行同源重組,通過抗性基因正向選擇突變株,pMAD引入顯色系統,通過反向選擇,更容易篩選突變株,亦可加入抗性標記;pKOR1質粒用于同源重組,克隆構建便捷,并通過secY反義RNA的表達來反向篩選突變株,敲除效率最高,且不需引入抗性基因,不改變宿主菌遺傳背景,是葡萄球菌基因敲除的理想選擇。
利益沖突:無
引用本文格式:武有聰,孟媛媛,丁百興,等.以質粒為基礎的同源重組技術在葡萄球菌基因敲除中的應用[J].中國人獸共患病學報,2019,35(7):580-586. DOI:10.3969/j.issn.1002-2694.2019.00.69
