2例先天性骨髓衰竭性疾病的診治分析
2019-07-01 08:44:32汪世花周朝陽楊國彪趙玲
醫藥前沿 2019年13期
汪世花 周朝陽 楊國彪 趙玲
(云南省楚雄州人民醫院血液科 云南 楚雄 675000)
先天性骨髓衰竭性疾病又稱先天性再生障礙性貧血,是遺傳性罕見疾病。該疾病臨床表現復雜,多于嬰幼兒起病,也有成年起病,常見有范可尼貧血(Fanconi anemia,FA)、先天性角化不良(Dyskeratosis congenital,DC)、Shwach man-Diamond綜合征(MDS)。先天性骨髓衰竭性疾病臨床易出現誤診及漏診,本文通過對2例確診病例進行臨床特征和診治進行分析,以提高對先天性骨髓衰竭性疾病的認識。
1.臨床資料
病例一:男,39歲,家族中無遺傳病史。反復鼻腔出血,左上肢六指畸形,皮膚色素沉著及咖啡、牛奶斑,小眼畸形(圖1)及聽力下降。外周血血小板減少;骨髓細胞學:骨髓增生活躍,巨核細胞數正常,血小板成熟障礙。予激素潑尼松片(1mg/kg·d)聯合達那唑6mg/d及免疫抑制劑(長春新堿1mg/w)、白介素-11連續治療無效,后開始有輕度貧血,為大細胞、大色素貧血,予補充葉酸、維生素B12治療仍無好轉。診斷考慮特發性血小板減少性紫癜,予治療后效果差。
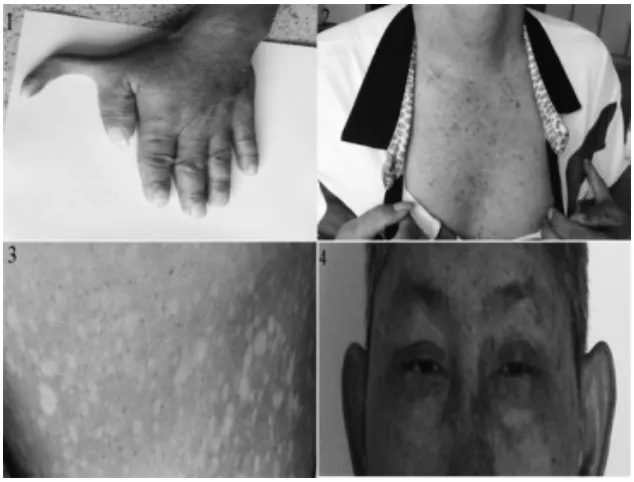
圖一:1左上肢六指畸形 2皮膚色素沉著 3皮膚咖啡牛奶斑 4小眼畸形及皮膚色素沉著
病例二:女,5歲10個月,家族中無遺傳病史,2歲6個月因視物不清診斷先天性青光眼、白內障,并行雙眼青光眼根治術。3歲9個月出現皮膚色素沉著,指、趾甲明顯變薄、脆裂、鄒縮等發育不良(如圖2)以及頭昏、乏力及全身多部位出血。血常規示:WBC1.94×109、L67.54%、HB 25g.L、PLT 14×109,骨髓細胞學:骨髓增生活躍,紅系以大紅細胞為主,血小板成熟障礙,考慮營養性巨幼細胞性貧血。但葉酸、維生素B12測定、遺傳代謝病氨基酸、酰基肉堿譜分析、尿液有機酸檢查及自身免疫疾病抗體16項及甲功全套均正常,上腹及盆腔CT檢查:肝臟、脾臟腫大。心臟彩超檢查:卵圓孔未閉。頭顱MRI檢查發現大腦發育不良及腦裂畸形,骨髓免疫分型檢查正常,高敏PNH檢查正常,白血病融合基因檢查均陰性,遺傳學檢查:5q-(-5/del 5q)、7q-(-7/del 7q)均為陰性,肝豆狀核變性相關血清銅、銅藍蛋白、尿銅檢查均正常。
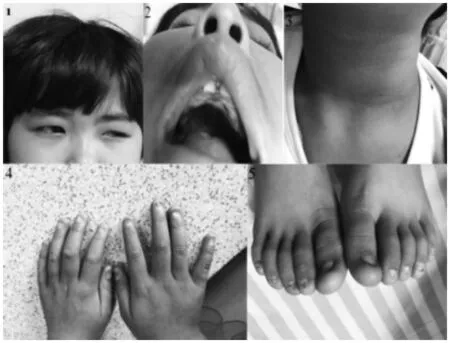
圖二:1青光眼術后,2口腔白斑,3皮膚色素沉著,4、5指、趾甲發育不良
2.實驗方法
2.1 染色體畸變檢測
例一通過在患者外周血中的淋巴細胞的培養樣本中加入誘變劑絲裂霉素C(MMC),觀察染色體自發斷裂、單體斷裂、裂隙等畸變情況,與沒有加入誘變劑的作對比。
2.2 基因突變檢測
例二采用PCR和基因測序的方法檢測標本中DKC1,TERC,TERT,NOP10,NHP2,TINF2,C16orf57,TCAB1八個基因編碼區全長及附近轉錄剪接位點的突變分析,檢測采用lon torrent PGM測序儀,共進行了53個擴增子的測序。
3.結果
3.1 染色體畸變
病例1通過外周血加入誘變劑觀察染色體檢查,MMC誘導染色體分析細胞數100個,MMC誘導畸變細胞數14個(正常0~4個)、MMC誘導畸變率14%(正常小于9%)、染色單體型畸變率14%(正常小于0~2%),確診FA。
3.2 基因突變
病例2先天性角化不良基因檢查,結果TINF2突變(c.845G>A,造成氨基酸的改變p.Arg282His),確診DC。
4.討論
遺傳性再生障礙性貧血,遺傳性全血細胞減少,癌前綜合征,遺傳性骨髓衰竭綜合征,染色體斷裂綜合征,是最常見先天性的骨髓衰竭綜合征的主要類型之一。人群中突變基因攜帶頻率為1/300,在高發人群與地區突變基因攜帶頻率可高達1/90[1],其發病率1/107~5/107。不同種族、不同人群發病率有所不同,該病平均診斷年齡為8歲(2~15歲),平均壽命為24.7歲(0~50歲)[2]。
該病有多臟器與多系統受累,屬于罕見疾病,診斷技術特別,FA的發展研究明顯慢于其他學科,因此對FA基本的認識缺乏。FA因為病因復雜,臨床表現多樣,如病例1,成年發病,但起病時外周血僅血小板減少,并沒有表現出FA典型的骨髓衰竭。根據骨髓細胞學檢查診斷特發性血小板減少性紫癜,治療效果差,結合患者軀體畸形、聽力下降等特殊體征,經染色體斷裂點檢查才最后確診,極大地影響臨床診斷與治療。根據國際范可尼研究中心總結了有關FA臨床診斷的主要及次要指征[3],染色體斷裂檢測被譽為診斷“黃金標準”。近年來基因檢測也已用于FA的診斷及分型,目前已發現16種致病基因(FANCA、B、C、D1、D2、E、F、G、I、J、L、M、N、O、P、Q),表現出相應的 FA 疾病類型,FA患者臨床表現的異質性可能與患者不同的突變類型及攜帶的突變基因有關。
DC也是最常見先天性的骨髓衰竭綜合征的主要類型之一,是人類發現的第一種端粒不穩定性疾病。導致DC的各種端粒酶相關基因突變,主要包括DKC1,TERC,TERT,NOPl0,NHP2,TINF2,POTlb,TCAB1等八個基因,另有40%的致病基因尚未明確。這些基因一般為X染色體隱性、常染色體顯性和常染色體隱性等3種遺傳方式。該病臨床表現復雜多樣,多累及中胚層及外胚層,其中皮膚粘膜病變最為典型,表現為皮膚色素沉著,指和趾甲營養不良及口腔粘膜白斑,病例2首先三個典型體征,其次為血液系統,表現為不同程度全血細胞減少,是本病死亡的主要原因,再次為其他系統累及,包括眼、耳、心血管、消化、呼吸、神經、內分泌等系統。該患兒2歲6個月就患青光眼,并累及心臟及神經系統,表現為先天性心臟病、大腦發育不良及腦裂畸形,輾轉多家醫院經血液系統骨髓形態、免疫、基因、染色體及遺傳代謝性疾病相關檢查未能確診,最后經TINF2基因突變確診為先天性角化不良。TINF2基因定位于14q11.2,編碼一種TIN2蛋白,此蛋白屬于編碼端粒酶的RNA組分基因相關保護復合體的一部分,其突變極大地減弱了端粒酶復合體延長端粒的功能,最終導致端粒的過度破壞及造血干細胞加速衰老,同時導致治療中對常規劑量造血干細胞移植預處理方案無法耐受。
先天性骨髓衰竭性疾病,治療方面尚無理想方案,異基因造血干細胞移植(allo-HSCT)仍為目前唯一治愈本病的手段,但allo-HSCT并不能改變患者成年后易患惡性疾病的傾向,尤其DC患者。此外,因先天性骨髓衰竭性疾病發病率低,報道較少,而且臨床表現多樣性,因此診斷困難,通過對臨床體征的仔細觀察及結合染色體畸變和基因檢查可以早期診斷,避免出現誤診及漏診。
