灰黃霉素超飽和體系及其共晶的跨膜滲透性研究
2019-05-16 01:40:46胡志遠陳瑩嫻劉曉慧王凱茹朱明強趙興華
中國獸醫雜志 2019年11期
胡志遠 , 陳瑩嫻 , 劉曉慧 , 王凱茹 , 朱明強 , 劉 靜 , 趙興華 , 何 欣
(1.河北農業大學動物醫學院 , 河北 保定 071000 ; 2.河北保定海關 , 河北 保定 071001)
灰黃霉素(Griseofulvin, Gri) 是1939年從青霉菌(Penicilliumspp.)培養液中得到的一種含氯代謝產物,用于治療皮膚真菌病,對毛發癬菌、小孢子菌、表皮癬菌等淺部真菌有良好的抗菌作用[1]。灰黃霉素在生物藥劑學分類系統(Biopharmaceutics classification system, BCS)中屬于低溶解性高滲透性的Ⅱ類藥物[2],口服生物利用度由藥物在胃腸道中的溶解度決定,由于灰黃霉素的溶解度極低,其在胃腸道中的溶解速率很小,口服生物利用度低,大大限制了其臨床應用。增加BCSⅡ類藥物的溶解度和溶出度是科研工作者所面臨的重要挑戰之一。目前,通常采用包括納米技術、水溶性載體、微粉化技術、無定形體、溶劑化物、固體分散體等技術提高難溶性藥物的溶解度,但是這些方法都有其應用的局限性[3-4],例如,納米混懸和固體分散技術存在穩定性差,制劑在放置過程中會出現溶出速率下降的問題。共晶是指藥物分子與共晶形成物以一定的比例,通過分子間非共價相互作用力形成的晶體。通過將藥物制備成共晶可以改變藥物的流動性、穩定性、溶解度和溶出速率甚至口感。
羥丙甲纖維素醋酸琥珀酸酯(Hydroxypropylcellulose acetate succinate, HPMCAS-HF)是羥丙甲纖維素的醋酸酯和琥珀酸酯混合物,有研究顯示,該纖維素類聚合物具有最優良的抑制藥物結晶的效應[5-6]。本研究在制備灰黃霉素共晶的基礎上,通過對添加HPMCAS-HF的灰黃霉素液態超飽和溶液、灰黃霉素-安賽蜜共晶以及灰黃霉素粉末的跨膜滲透性進行比較分析,為研究灰黃霉素的藥動學提供科學依據。
1 材料與設備
灰黃霉素(Griseofulvin,Gri):廣東臺城制藥有限公司;安賽蜜鉀(Acesulfame potassium,AceK):Tokyo Chemistry Industry co., Ltd., Japan;羥丙甲纖維素醋酸琥珀酸酯(HPMCAS-HF):大連業建貿易有限公司;X-射線衍射儀:Bruker Axsinc, Madison, Wisconsin;紫外-可見光分光光度計:UV-2450,Shimadzu,Japan;HC-188透皮擴散儀:天津市正通科技有限公司;透析膜(截留分子量為8 000-14 000):用10%的NaHCO3,70 ℃水浴20 min以除去硫化物;10 mM的EDTA 70 ℃ 水浴20 min以除去重金屬;去離子水70 ℃ 水浴20 min以除去甘油;其他試劑均為分析純。
2 試驗方法
2.1 灰黃霉素紫外分光光度計測定標準曲線的建立 按照文獻[7]的方法建立灰黃霉素的紫外分光光度計的測定方法。
2.2 共晶的制備 利用離子交換法[8]制備灰黃霉素-安賽蜜共晶。稱取灰黃霉素7.1 g和安賽蜜鉀4.02 g加入到甲醇和水的水溶液中,加入適量的鹽酸,磁力攪拌器攪拌24 h,取濾渣放置于60 ℃烘箱中干燥6 h。
2.3 灰黃霉素-安賽蜜共晶的X射線衍射表征 分別取灰黃霉素和灰黃霉素-安賽蜜共晶樣品適量,將樣品平鋪,電壓45 kv,電流40 mA,掃描步長0.02°,以0.02°/s的速度在 (5°~35°)掃描,繪制X射線衍射圖。
2.4 灰黃霉素以及灰黃霉素-安賽蜜共晶的HPMCAS-HF溶液的配制 配置適當濃度的HPMCAS-HF溶液。將適量的灰黃霉素和灰黃霉素-安賽蜜共晶溶解在上述溶液中以達到最大的濃度。
2.5 溶液的平衡溶解度的測定 分別選取灰黃霉素原料藥和灰黃霉素-安賽蜜共晶樣品適量,加入到10 mL水中和HPMCAS-HF的緩沖溶液中,密封,37 ℃,溶液在過飽和狀態下攪拌72 h,溶液達到平衡后,10 000 r/min離心15 min取上清液,取濾液用紫外分光光度計測定濃度。
2.6 體外擴散率的測定 利用Fran’z擴散池進行藥物的體外擴散試驗。池口的有效擴散面積為0.785 cm2,上下兩個半池分別為擴散池和接受池,接受池的容積為5 mL。將10、8、5、2 μg/mL的灰黃霉素水溶液和165 μg/mL、82.5 μg/mL、55.27 μg/mL的灰黃霉素的0.1%的HPMCAS-HF溶液以及14 μg/mL灰黃霉素-安賽蜜共晶水溶液,65.60 μg/mL共晶的HPMCAS-HF溶液加入Fran’z透皮儀的樣品池,接收液為去離子水。在接受池和擴散池中間移入處理好的透析膜,夾緊后接受池中加入去離子水,擴散池中加入待試藥液2.5 mL。然后置于(37±0.03)℃的循環水浴中,接受池轉子攪拌速度為400 r/min,加入樣品后,分別于設定的時間間隔:30、45、60、90、120、180、240、300、360 min 由取樣口取1 mL接受液,同時補充等體積的去離子水。將不同時間取出的接受液樣品用上述方法進行吸光度的測定,代入回歸方程[9]。并計算穩態滲透速率。
式中:C是接受液的校正濃度(μmol/L),Cn是第n個取樣點接受液樣品的實測濃度(μg/mL),∑Ci是第n個取樣點前實測濃度之和,V 是接受室體積(即5 mL),V0是每次取樣的體積(即1 mL)。
體外滲透速率按Fick’s 擴散方程(2)計算: Jss=V/A×dc/dt(2)
式中:Jss是穩態滲透速率(μg/cm2·h),V是接受室體積(即5 mL),A 是皮膚有效擴散面積(即3.14×0.52cm2),c 是接受室的校正濃度(μg/mL),t是時間(h),dc/dt是接受液濃度與時間曲線的直線部分的斜率。
3 結果與分析
3.1 灰黃霉素標準曲線的繪制 選用292 nm 為測定波長,經回歸得標準曲線方程為y=0.097X+0.007 6,R2=0.999 9。在0~20 μg/mL范圍內線性關系良好。如下表1和圖1。
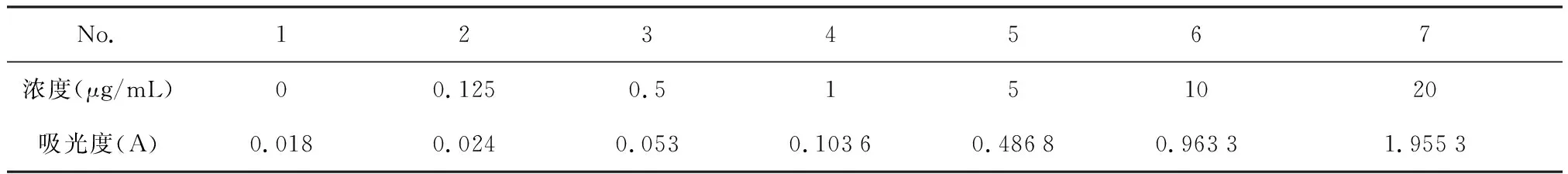
表1 不同濃度灰黃霉素的吸光度
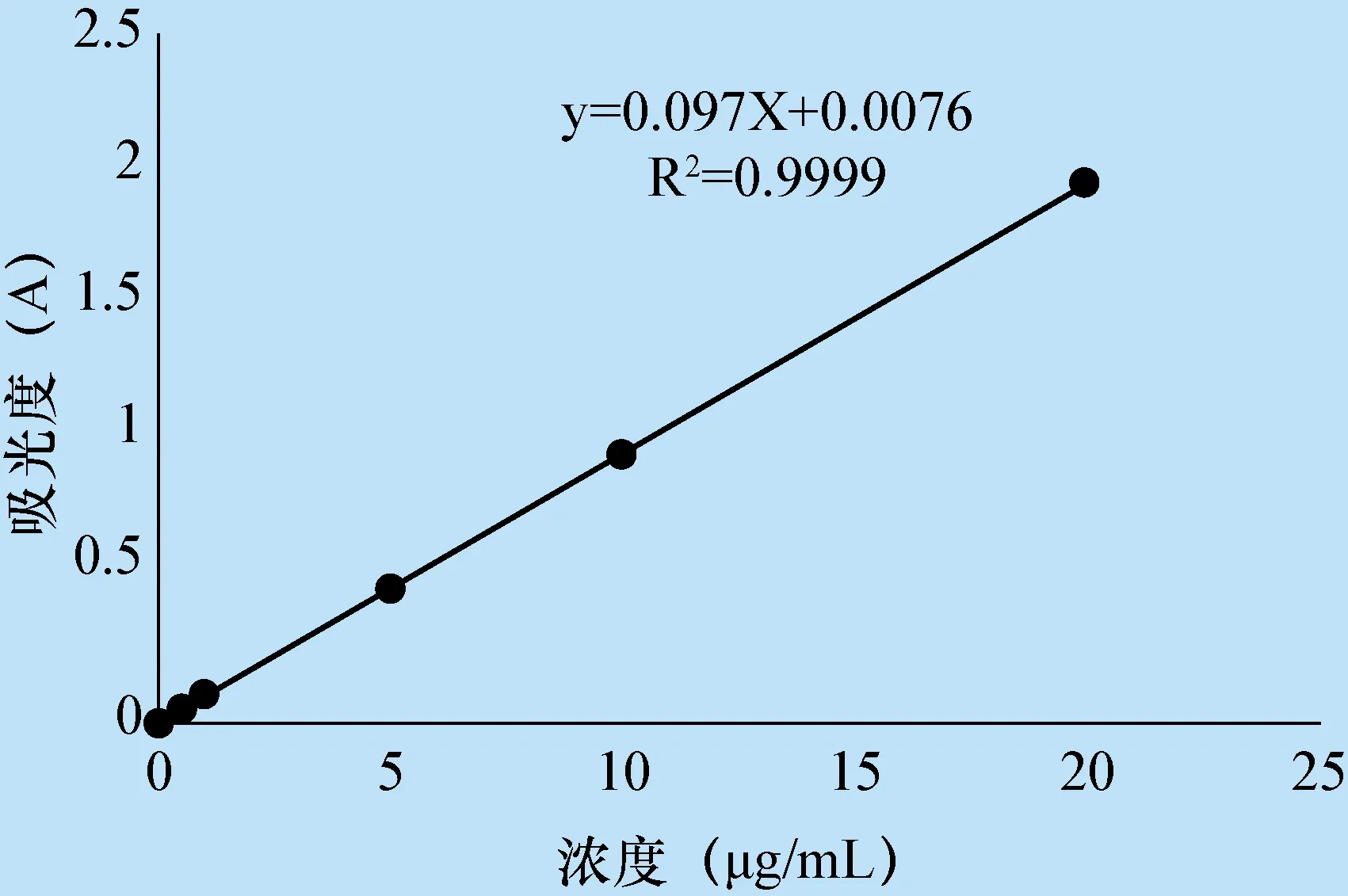
圖1 灰黃霉素的紫外標準曲線
3.2 灰黃霉素的平衡溶解度 如表2所示,灰黃霉素添加聚合物之后可以顯著提高灰黃霉素的溶解度。灰黃霉素-安賽蜜共晶的溶解度在添加聚合物HPMCAS-HF之后也有顯著提高。

表2 不同灰黃霉素溶液的溶解度 (37 ℃)
3.3 灰黃霉素-安賽蜜共晶的X射線衍射圖 試驗樣品的X射線衍射(PXRD)分析結果如中插彩版圖2所示,利用離子交換法制備的灰黃霉素-安賽蜜共晶與文獻中利用研磨法制備的共晶出現了相同的PXRD譜圖的特征峰,用研磨的方法(AceH gri G)和溶劑法(AceH gri S 和AceK gri S)制備的灰黃霉素-安賽蜜共晶的PXRD圖與文獻報道(paper data)的一致[7]。
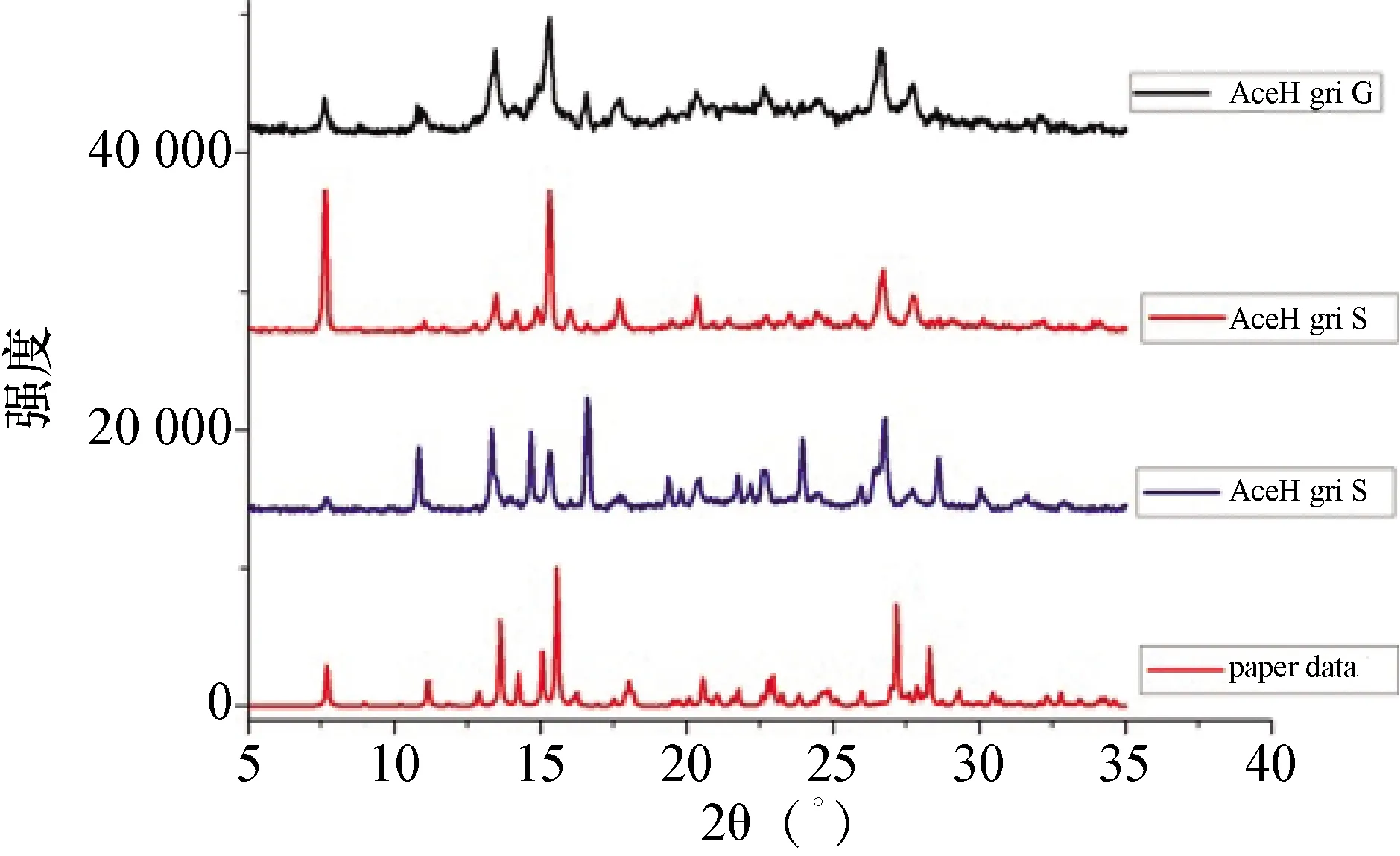
圖2 灰黃霉素-安賽蜜共晶的PXRD圖
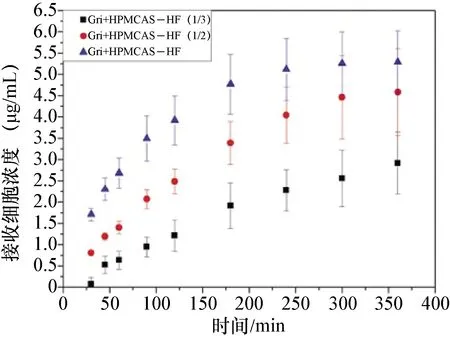
圖3 超飽和灰黃霉素的跨膜滲透曲線
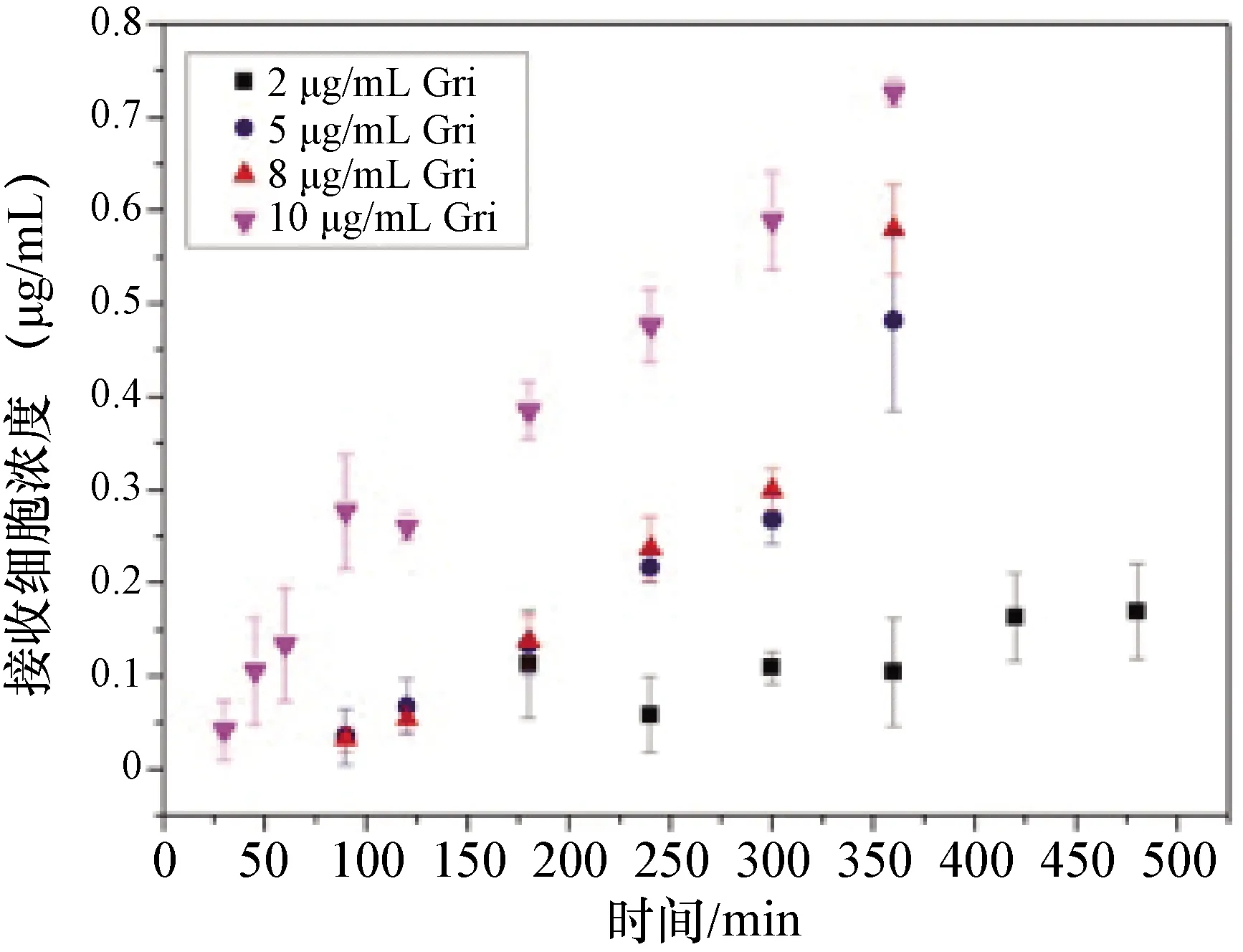
圖4 低于飽和溶解度時灰黃霉素的跨膜滲透曲線
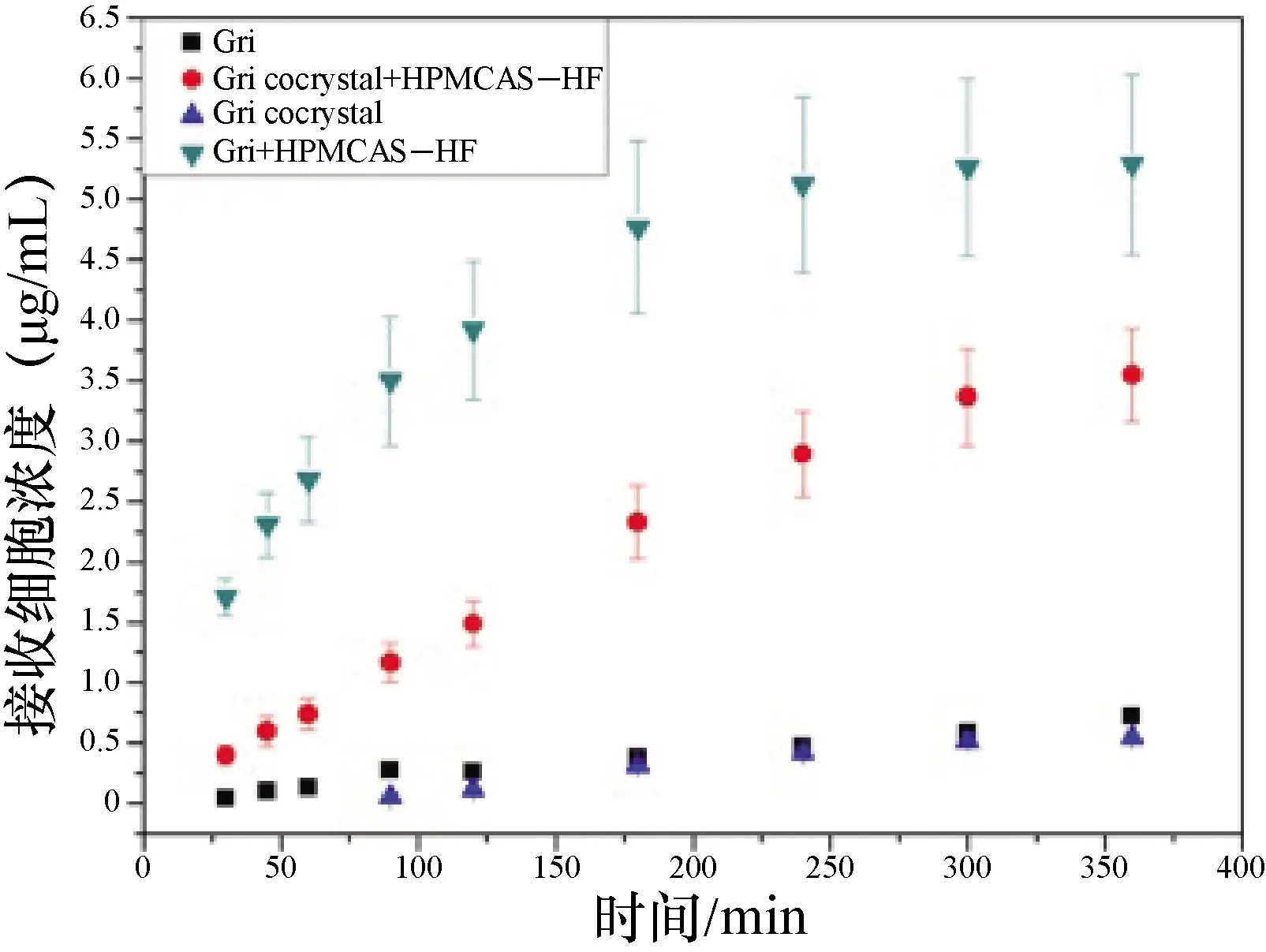
圖5 灰黃霉素添加聚合物前后的跨膜滲透曲線
3.4 灰黃霉素的擴膜滲透性及穩態滲透速率 不同物質跨膜滲透的穩態滲透速率如下表3。由中插彩版圖3-5跨膜滲透曲線和表3的穩態滲透速率數據可以看出,低于灰黃霉素飽和溶解度的樣品的跨膜滲透速率均較低(中插彩版圖4),其中5 μg/mL和10 μg/mL的跨膜滲透速率只有0.006 37 μg/cm2·h。制備的灰黃霉素-安賽蜜共晶在一定程度上增加了藥物的溶解度,其在HPMCAS-HF中的溶解度也有提高,穩態滲透速率也相應地提高了(中插彩版圖5)。但是灰黃霉素在HPMCAS-HF中處于超飽和狀態時,具有最高的穩態滲透速率(中插彩版圖4),為0.153 μg/cm2·h,比灰黃霉素在水中提高了12倍,比灰黃霉素共晶在水中提高了11.7倍,比灰黃霉素共晶在HPMCAS-HF中提高了2倍。

表3 不同物質跨膜滲透的穩態滲透速率
4 討論
藥物制劑技術的發展提高了新藥開發的成功率,許多藥物存在諸如水溶性差和生物利用度低等缺點,研究人員常借助于制備藥物的無定形態以改善藥物的溶解度和溶出速度,但無定形態藥物在水溶液中能否形成超飽和溶液是其提高溶解性的關鍵。從熱力學角度來說,從超飽和溶液中獲得的溶解度的提升與添加諸如表面活性劑的提升是不一樣的。超飽和溶液的化學能和熱力學性質比飽和溶液和亞飽和溶液的要高。添加物增加濃度是增加平衡溶解度而不是增加化學勢能。例如將藥物添加在含有表面活性劑的微團中,可以增加溶解度,但不增加“自由藥物”的量。自由藥物的數量與溶液的熱力學活性有關,這個參數還決定跨膜的程度。這個區別很重要,因為這樣的超飽和溶液就可以增加物質的跨膜轉運能力[10-11]。另外,超飽和溶液會形成一個非結晶的藥物富集相。聚合物與無定形態結合形成的固態分散體也是借助于聚合物能夠維持其穩定性的作用,特別是在溶液中形成超飽和的能力。無定型的固體分散體由于具有較高的溶解度和溶出速率而出現超飽和溶液,一定量的聚合物可以通過抑制結晶的形成而使這個藥物富集相以納米級的框架形成[12-13],從而增加藥物的跨膜轉運。
本試驗以灰黃霉素為例,此前已有文獻報道了它的無定形態在水溶液中會發生快速結晶而導致它失去超飽和的優勢,測得無定形態與結晶態的溶出速度沒有提高,另外制得的共晶能夠達到藥物最大濃度的3倍左右,但共晶在水溶液中也存在著藥物分子去飽和析出的問題,即也無法維持藥物的超飽和狀態。Lynne等[14]證實了在眾多聚合物中,HPMC-AS-HF是一種能夠有效的結晶抑制劑,在本試驗中我們首先通過離子置換法制得了高濃度的灰黃霉素溶液,試驗結果表明,加入0.1%的HPMC-AS-HF能夠長時間維持溶液中灰黃霉素的濃度至165 μg/mL,這一超飽和溶液長時間維持了藥物較高的跨膜滲透速率,灰黃霉素的HPMCAS-HF的超飽和溶液的滲透速率為0.153 μg/cm2/h,遠遠高于灰黃霉素原液的滲透速率,大約是其12.05倍。表明這一方法極為有效的提高了溶液中藥物的游離態濃度,這一方法較加入β環糊精、表面活性劑和PEG等助溶手段而言具有較大優勢,因為一般的助溶手段均不能增加游離態藥物的濃度,因此常常出現藥物表觀濃度增加但跨膜滲透速度并未發生變化。在本研究中,共晶的制備并沒有增加藥物的滲透性,但是添加聚合物之后的共晶溶液的滲透性卻有了顯著的增加。這為本課題組將進行灰黃霉素、灰黃霉素共晶以及灰黃霉素的聚合物溶液的生物利用度研究,提供了重要的理論依據。
