活性炭吸附-碘量法測低品位礦石中金的不確定度評定
2019-05-11 03:00:06楊海江單召勇
世界有色金屬 2019年4期
楊海江,單召勇
(山東黃金冶煉有限公司,山東 萊州 261400)
關鍵字:礦石;金;活性炭吸附-碘量法;標液;不確定度
在黃金礦產的勘探、開采、浮選及氰化過程中,常常伴隨有低品位樣品,對其金含量進行測定有助于對礦產資源儲量和開采價值進行評定,及對浮選或氰化指標進行考核。目前常用的檢測方法有兩種,即活性炭吸附-碘量法和AAS法,而碘量法由于檢測成本低、使用時間長而廣泛使用。
測量不確定度合理地表征被測量值的分散性,是與測量結果相聯系的參數,是評價測量結果的重要指標。礦石中金的分析不確定度評定,許多文獻早有報道,但都只針對含金量較高的礦石樣品,低品位礦石樣品中金的測量不確定度評定則鮮有涉及。
本文通過對活性炭吸附-碘量法測量低品位礦石中金的不確定度來源進行分析,并對其不確定度分量進行量化計算,找出影響檢測結果的主要因素,對今后優化低品位礦石中金含量測定方法有一定的借鑒意義。
1 測量方法
1.1 基本原理
試樣經反王水處理,用王水溶樣,活性炭吸附富集后,經碳化、灰化、王水溶解、水浴蒸酸后,在乙酸介質中,加入氟化氫銨、EDTA掩蔽少量的鐵、銅等雜質,碘化鉀與三價金反應置換出等當量的碘單質,以淀粉為指示劑,用硫代硫酸鈉標準滴定液滴定。主要反應為[1]:
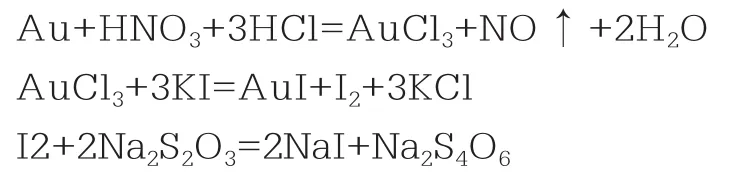
1.2 測定步驟
準確稱取低品位礦石樣品30.00g于400mL燒杯中,用水潤濕試樣,用新配制的反王水(1+1)處理樣品,至無紅棕色氣體溢出,加入新配制的王水(1+1)至160mL左右,加蓋表面皿,煮沸30~40min,至體積小于50 mL時取下,用水洗滌表面皿和杯壁稀釋至120 mL左右,放冷后用活性炭吸附富集,經碳化、灰化后,加入1~3滴飽和氯化鈉溶液,用王水溶解,于水浴鍋上蒸干,用鹽酸趕硝2次,加入少量醋酸溶液、氟化氫銨、EDTA、碘化鉀,以淀粉為指示劑,用硫代硫酸鈉標準滴定液滴定至溶液由藍色轉變為無色即為終點。同時,以金標液標定硫代硫酸鈉,計算其滴定度[2]。
1.3 計算公式
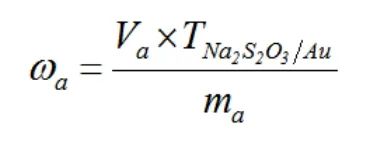
式中:ωa為試樣中金含量(g/t),Va為樣品消耗的硫代硫酸鈉標液的體積(mL),為硫代硫酸鈉對金的滴定度(ug/mL),ma為試樣質量(g)。
2 不確定度評定
2.1 不確定度的來源
不確定度的來源主要包括樣品稱量質量、滴定液消耗體積、標定硫代硫酸鈉的滴定度和重復性測定等[3]。
2.2 不確定度分量的分析及量化
2.2.1 樣品稱量質量的不確定度
本實驗采用梅特勒生產的型號為PL402-L的電子天平進行稱量,根據鑒定證書顯示重復性誤差為±0.01g,按照均勻分布,稱樣量為30.00g,稱樣時清零的不確定度也計算一次,則稱樣過程產生的不確定度為:稱量質量的合成標準不確定度為:
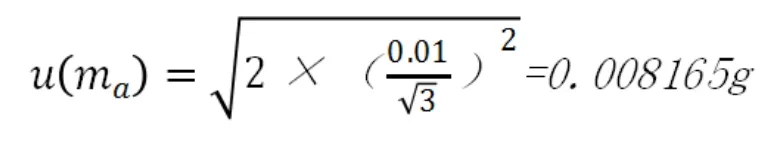
相對合成標準不確定度為:
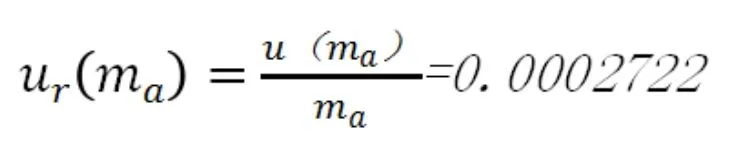
2.2.2 滴定液消耗體積的不確定度
滴定樣品消耗硫代硫酸鈉標準滴定溶液體積的不確定度主要來源于讀數誤差、滴定管校準偏差、環境溫度變化引起的偏差。
(1)讀數誤差的不確定度
50mL滴定管的分度值為0.1mL,估讀誤差為0.05ml,按照均勻分布,不確定度為:
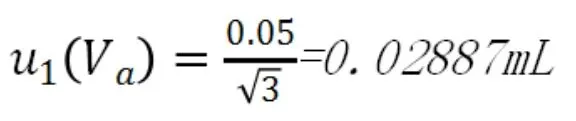
(2)滴定管校準的不確定度
根據JJG-196-1999,A級滴定管的最大誤差為±0.05mL,按照均勻分布,不確定度為:
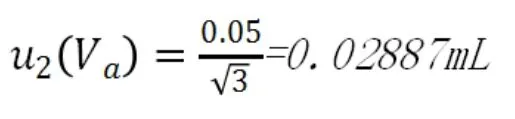
(3)環境溫度變化導致的不確定度
滴定管的校準溫度為20℃,實際使用過程中室溫會有±5℃的變化,水的膨脹系數為2.1×10-4/℃,按照均勻分布,實驗中消耗的硫代硫酸鈉標準溶液體積為Va=0.67mL,不確定度為:
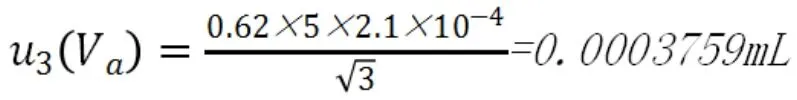
d.合成標準不確定度
綜上,則滴定液消耗體積的合成不確定度為:
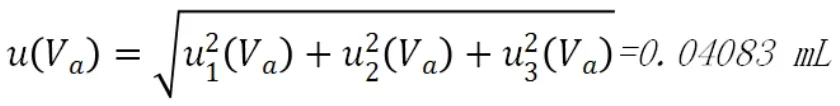
相對標準不確定度為:
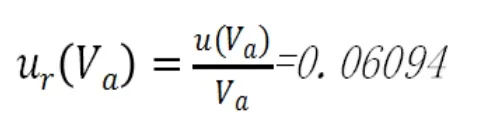
2.2.3 硫代硫酸鈉滴定度的不確定度
硫代硫酸鈉標準滴定液的滴定度的標定過程為,用移液管吸取10mL濃度為100ug/mL金標準溶液,用硫代硫酸鈉標準溶液進行滴定,按照下式計算其滴定度:
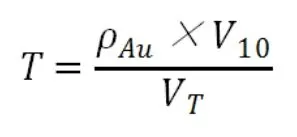
式中:T為硫代硫酸鈉標準滴定溶液對金的滴定度(ug/mL),V10為10mL移液管移取金標液的體積,VT為標定過程中消耗的硫代硫酸鈉標準滴定溶液的體積。
可見,標定過程中不確定度的主要來源為金標準溶液的濃度、移液管吸取標液的體積、消耗硫代硫酸鈉標液的體積和重復標定。
(1)金標準溶液濃度的不確定度
金標準溶液的制備過程為用電子天平稱取0.1000g純度為99.99%的金標溶解后定容至1000mL的容量瓶中。
a.金標稱量
電子天平為梅特勒的XP204可讀性為0.1mg,則區間半寬為0.05mg,最大允許誤差為±0.2mg,稱樣清零時計算一次不確定度,按照均勻分布,其不確定為:
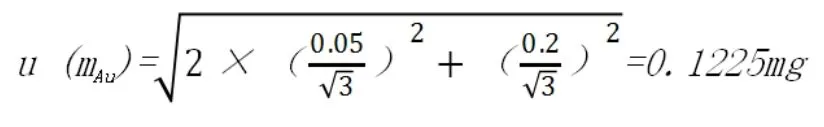
相對標準不確定度為:
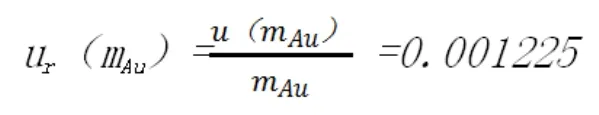
b.金標純度
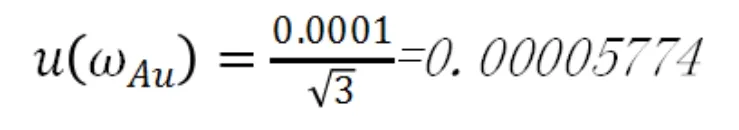
相對標準不確定度為:
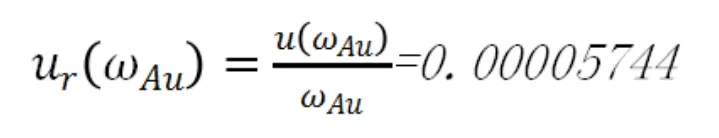
c.定容誤差
A級1000mL容量瓶的允許誤差為±0.4mL[4],按照均勻分布,由校準引起的標準不確定度為:
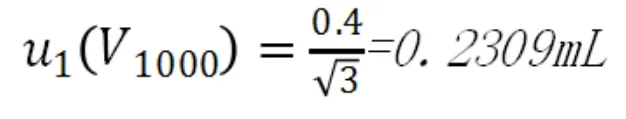
校準溫度為20℃,按照室溫±5℃的變動,由溫度引起的不確定度為:
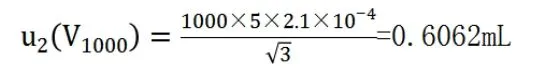
則,金標定容過程的合成標準不確定度為:
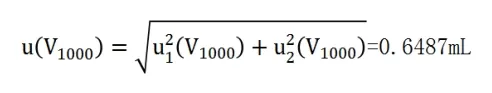
其相對標準不確定度為:
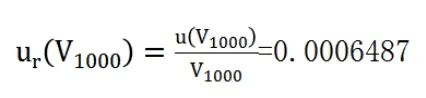
d.合成標準不確定度
綜上,金標準溶液濃度的相對合成標準不確定度為:
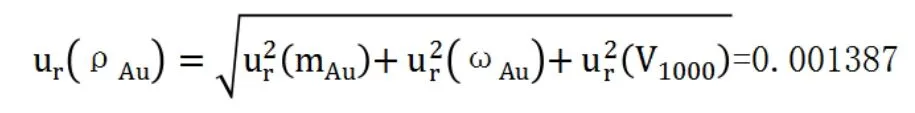
金標準溶液濃度ρAu為100ug/mL,則其標準不確定度為:
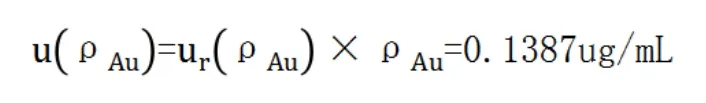
(2)移液管吸取標液體積的不確定度
用移液管吸取金標液過程中的不確定度主要來源有3方面∶A級10mL移液管的允許誤差為±0.02mL,人員重復性操作示值誤差為±0.01mL,室溫與校正時溫度存在±5℃變動,按照均勻分布,計算其各分量的標準不確定度如表1所示。

表1 移液管吸取標液過程中的不確定度分量
則移液管吸取金標液的體積合成標準不確定度為:
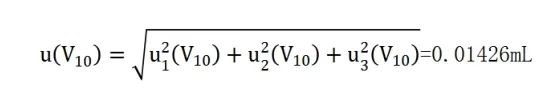
相對標準不確定度為:
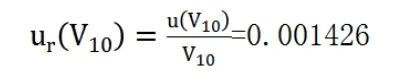
(3)標定過程消耗標液體積的不確定度
關于滴定管使用過程中不確定度的來源及計算參見

相對標準不確定度為:
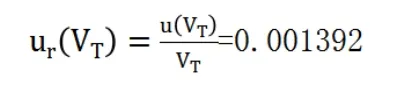
(4)重復標定引入的不確定度
重復標定過程中消耗標液的體積為32.60、32.55、32.57、32.55mL,用貝塞爾公式[5]計算不確定度和相對標準不確定度如表2:
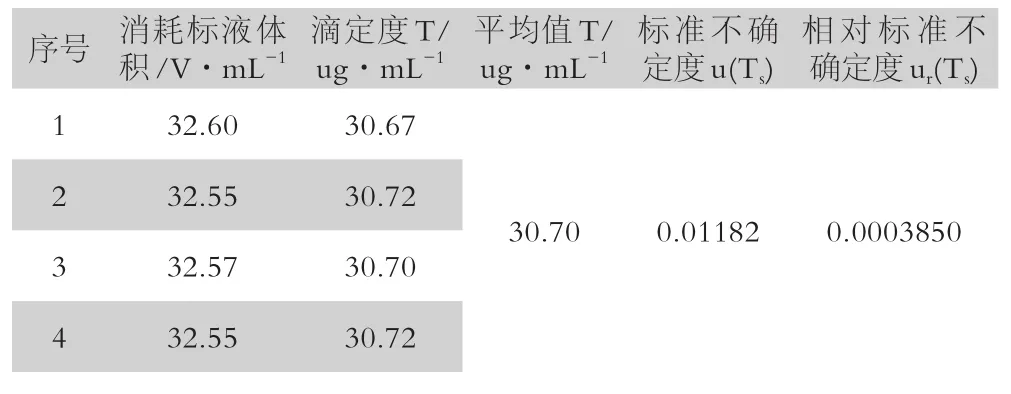
表2 重復標定引入的不確定度
綜上,則硫代硫酸鈉滴定度的相對合成標準不確定度為:

2.2.4 重復性測定引入的不確定度
對樣品由3人進行了10次測定[6],用貝塞爾公式計算不確定度和相對標準不確定度如表3:
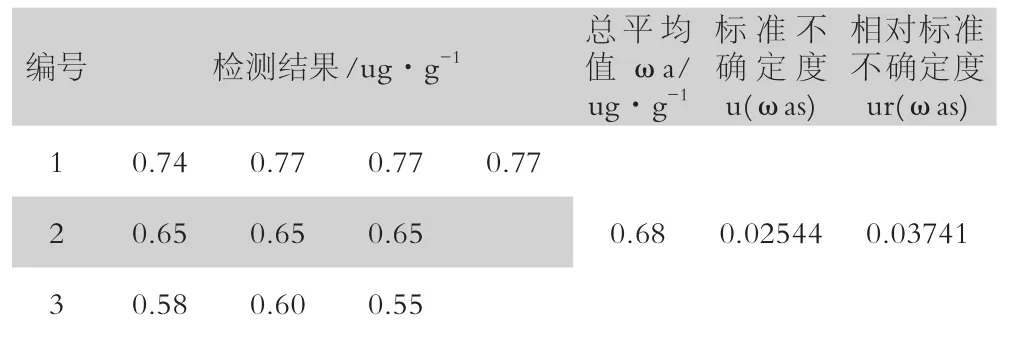
表3 碘量法重復性測定引入的不確定度
2.3 不確定度分量匯總
將活性炭吸附-碘量法中的不確定度分量匯總如表4所示∶

表4 活性炭吸附-碘量法中的不確定度分量匯總

重復標定 30.70ug/mL u(Ts) 0.01182 0.0003850重復性測定 0.68ug/g u(ωas) 0.02413 0.03548
2.4 合成標準不確定度
根據式1.1.3,活性炭吸附-碘量法測金的相對合成標準不確定度為:
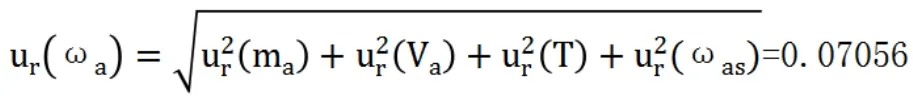
測定樣品中金含量的標準不確定度為:
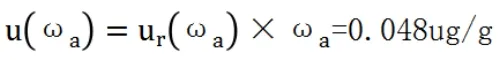
2.5 擴展不確定度和測定結果表示
當自由度未知時,在95%的置信概率下,包含因子k0.95=2,測定結果的擴展不確定度為[7]:
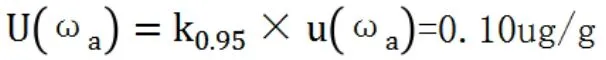
故用活性炭吸附-碘量法測定該樣品的結果可表示為0.68±0.10ug/g,即該樣品中的金含量有95%的可能性在0.58ug/g~0.78ug/g之間。
3 結論
采用活性炭吸附-碘量法測定樣品中金含量為0.68±0.10ug/g,擴展不確定度UP=0.10ug/g。活性炭吸附-碘量法的不確定度主要來源為樣品滴定過程消耗標液體積和重復性測定,滴定度標定和樣品稱量過程引入的不確定很小。造成這種結果的主要原因有:
(1)試樣含金量太低,在滴定過程中消耗標液體積很小,使用滴定管讀數的相對誤差較大;
(2)硫代硫酸鈉標準溶液濃度偏高;
(3)不同操作者在滴定終點判斷過程存在偏差,但由于消耗標液體積很小,其造成的相對偏差較大;
(4)試樣含金量太低,各種干擾元素相對較高,經常出現紅終點、黃終點等現象,影響滴定終點判斷。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
理化檢驗-化學分冊(2020年5期)2020-06-15 11:36:00
中國金屬通報(2019年7期)2019-08-13 07:44:34
當代陜西(2019年8期)2019-05-09 02:22:48
——硫代硫酸鈉
數理化解題研究(2019年7期)2019-03-27 06:03:08
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
山東化工(2017年7期)2017-09-16 05:19:25
專用汽車(2016年4期)2016-03-01 04:13:43
高中生學習·高三版(2015年2期)2016-01-04 22:41:24
