組合策略促進堿性果膠酶在畢赤酵母中高效表達
2019-04-25 07:29:02陳雙全堵國成
食品與生物技術學報 2019年2期
陳雙全 ,劉 松 ,堵國成 *,陳 堅
(1.江南大學 工業生物技術教育部重點實驗室,江蘇 無錫 214122;2.江南大學 生物工程學院,江蘇 無錫214122)
堿性果膠酶(EC 4.2.2.2,PGL)可在堿性條件下將果膠質裂解為不飽和的寡聚半乳糖醛酸,廣泛應用于紡織、食品、造紙及環境領域[1-2]。為實現高效生產,PGL已在枯草芽孢桿菌[3]、大腸桿菌[4]及畢赤酵母(P.pastoris)[5]中表達。其中,重組大腸桿菌獲得的PGL產量最高,達到4 478 U/mL[4]。然而,大腸桿菌表達PGL仍存在諸多問題,包括培養基成本高(蛋白胨類)、需添加價格昂貴的誘導劑(異丙基-β-d-硫代半乳糖苷)及易感染噬菌體等。對于工業生產,P.pastoris表達系統具有如下優勢:目標基因整合于基因組表達,穩定性高;營養需求低;易于高密度培養;分泌系統強,胞外雜蛋白極少,目標蛋白易于分離純化[6]。本團隊前期研究中,成功實現了PGL在P.pastoris中的表達,產量達到863 U/mL,經補料策略優化可進一步提高[7]。
大量研究表明,外源蛋白在P.pastoris中高效表達受一系列因素影響[8-10]。首先,目標基因拷貝數將在基因轉錄水平上影響外源蛋白在P.pastoris中的表達[11]。其次,P.pastoris對密碼子顯示出偏好性也將在翻譯階段調控基因表達[12-13]。此外,外源蛋白的過量表達會引起未折疊蛋白效應(UPR),對外源蛋白的折疊和轉運有重要作用。UPR包括大量的分泌輔助因子或分子伴侶,如:轉錄因子(HAC1)、蛋白二硫鍵折疊酶(PDI)、內質網中蛋白折疊氧化還原輔助因子(ERO1)、結合蛋白(BIP)、泛素共軛酶(UBC1)以及輔助轉運及分泌過程的其他分子伴侶等[14-16]。因此,對基因拷貝數、密碼子偏好性及UPR等因素的調控是促進PGL在P.pastoris中表達的重要策略。
本團隊在前期研究中篩選得到的一株產PGL的Bacillussp.WSHB04-02菌株,經分子改造提高了其熱穩定性及催化活性[17]。本研究以PGL高熱穩定性突變體K314M為研究對象,通過密碼子優化及過量表達分子伴侶ERO1及UBC1,以期提高PGL在P.pastoris中的分泌表達效率,并在3 L罐水平上考查了重組菌的發酵性能。
1 材料與方法
1.1 材料
1.1.1 菌株與質粒編碼Bacillussp.WSHB04-02 PGL高熱穩定性突變體K314M基因的載體pET-22b(+)/pgl,由本研究室在前期工作中構建[17]。 表達載體pPIC9K及pGAPZA,表達宿主P.pastorisGS115(his-)及克隆宿主E.coliTOP10均購于Invitrogen公司(美國)。
1.1.2 主要試劑限制性內切酶EcoR I、NotI、SacI、BamH I、Bsp119 I、KpnI 和 AvrII 購 自ThermoFisher公司 (美國)。膠回收柱回收試劑盒、Prime STAR HS DNA聚合酶、酵母Total RNA提取試劑盒 (Yeast RNAiso Kit),反轉錄試劑盒(Prime Script RT reagent Kit with gDNA Eraser)及定量PCR試劑盒 (SYBR Premix Ex Taq TM Tli RNase H Plus)購自大連寶生物Takara公司。One Step Cloning Kit試劑盒購自南京諾唯贊生物科技有限公司。質粒提取試劑盒、G418、Zeocin、蛋白定量試劑購自生工生物工程(上海)有限公司。聚半乳糖醛酸購自Sigma公司(美國)。其他常用試劑及藥品均為分析純,進口分裝或國產。
1.1.3 培養基YPD培養基(g/L):蛋白胨20、酵母提取物10、葡萄糖20。
BMGY 培養基(g/L):蛋白胨 20、酵母提取物10、甘油 40、YNB13.4、生物素 0.000 4,0.1 M 磷酸鹽緩沖液,pH 6.0。
BMMY 培養基(g/L):蛋白胨 20、酵母提取物10、YNB13.4、 生物素 0.000 4,9%甲醇 (v/v),0.1 M磷酸鹽緩沖液,pH 6.0。
PTM1 溶 液 (g/L):CuSO4·5H2O 6.0、KI 0.08、MnSO4·H2O 3.0、Na2MoO4·2H2O 0.2、H3BO30.02、CoCl20.5、ZnCl220.0、FeSO4·7H2O 65.0、生物素 0.2,H2SO45.0 mL/L。
1.2 方法
1.2.1 PGL及分子伴侶表達載體構建
1)PGL表達載體pPIC9K-PGL構建 研究發現不同物種對密碼子使用頻率不同,P.pastoris對密碼子顯示出偏好性,也將在翻譯階段調控基因表達[12-13]。基于P.pastoris密碼子偏好性對PGL高熱穩定性突變體K314M的基因優化,并克隆至宿主pUC57(金斯瑞生物科技有限公司)。以pUC57-PGL為模板,用引物PGL-F/R(表1)擴增PGL基因,PCR擴增條件:95℃預變性5 min;隨后進行34個循環(95 ℃ 30 s,60 ℃ 30 s,72 ℃ 1.5 min), 最后 72 ℃保溫10 min。載體pPIC9K用EcoR I及NotI雙酶切,PCR片段及酶切后的載體柱回收,根據One Step Cloning Kit試劑盒說明書進行重組連接,連接產物轉化E.coli TOP10,質粒提出得到PGL表達載體pPIC9K-PGL。未經密碼子優化的K314M基因通過引物PGLN-F和PGLN-R從pET-22b (+)/pgl擴增得到,并克隆至pPIC9K中EcoR I-NotI位點,得到表達載體pPIC9K-PGLN。
2)內參基因載體pPIC9K-GAPDH構建 內參基因在各組織和細胞中表達相對恒定,常用做參照物,在測定基因轉錄水平及拷貝數時甘油醛-3-磷酸脫氫酶(GAPDH)(GenBank No:U62648.1)常用做內參基因[18-19],以P.pastorisGS115 cDNA為模板,用引物GAPDH-F/R(表1)擴增GAPDH基因,PCR擴增條件:95℃預變性5 min;隨后進行34個循環(95 ℃ 30 s,60 ℃ 30 s,72 ℃ 1 min), 最后 72 ℃保溫10 min。載體pPIC9K用EcoR I及NotI雙酶切,PCR片段及酶切后的載體柱回收,根據One Step Cloning Kit試劑盒說明書進行重組連接,得到GAPDH載體pPIC9K-GAPDH。
3)分子伴侶共表達載體的構建 以P.pastorisGS115 cDNA為模板,分別用引物ERO1-F/R和UBC1-F/R (表 1) 擴 增 ERO1 (GenBank No:XM_002489600.1) 和 UBC1 (GenBank No:XM_002493814.1)基因。PCR擴增條件為:95℃預變性 5 min,隨后進行34個循環(95℃ 30 s,60℃30 s,72℃ 1 min), 最后 72℃保溫 10 min。 載體pGAPZA用Bsp119 I及KpnI雙酶切,PCR片段及酶切后的載體柱回收,根據One Step Cloning Kit試劑盒說明書進行重組連接,分別得到ERO1和UBC1表達載體pGAPZA-ERO1及pGAPZAUBC1。利用BglII與BamH I為同尾酶效應,將構建好的pGAPZA-ERO1用BglII與BamH I雙酶切,膠回收得到完整的GAP-ERO1-AOX(TT)表達框,再與用BamH I單酶切的pGAPZA-UBC1載體連接,得到共表達ERO1和UBC1的載體pGAPZERO1-UBC1。

表1 本研究中所用PCR引物Table 1 Primers used for PCR in this study
1.2.2 P.pastoris GS115轉化與篩選為表達PGL基因,用SacI將質粒pPIC9K-PGL線性化,電轉化至感受態P.pastorisGS115菌株。轉化后涂布MD平板,30℃培養3~4 d,經MD/MM平板篩選Mut+/Muts菌株,再經YPD+1-4 mg/mL G418平板初步篩選基因拷貝數,挑選5~10個重組菌進行搖瓶發酵。
為共表達分子伴侶,用AvrII線性化pGAPZAERO1,pGAPZA-UBC1 及 pGAPZA-ERO1-UBC1,電轉化至以第1次轉化篩選得到的重組菌株GS115/PGL14#,涂布 YPD+100 μg/mL Zeocin 平板,30 ℃培養 3~4 d, 經 100~400 μg/mL Zeocin平板初步篩選基因拷貝數,挑選5~10個重組菌進行搖瓶發酵。
1.2.3 重組P.pastoris搖瓶發酵搖瓶培養條件:從固體培養基上挑取單菌落接種于YPD培養基中30℃,220 r/min培養14 h,以10%接種量轉接于BMGY培養基中30℃,220 r/min培養24 h,收集菌體,生理鹽水清洗2次,轉接至BMMY培養基中22℃,220 r/min誘導PGL表達,每24 h補加1.5%甲醇(v/v)。
細胞生長情況及酶活每24 h取樣測定,篩選得到的較優重組菌株通過熒光定量PCR準確測定其基因拷貝數。
1.2.4 3 L罐發酵從平板上挑取單菌落接種于YPD培養基中30℃,220 r/min培養24 h,以10%接種量接種于包含800 mL分批發酵培養基(85%磷酸 26.7 mL/L,CaSO40.93 g/L,K2SO418.2 g/L,MgSO4·7H2O 14.9 g/L,KOH 4.13 g/L,甘油 40.0 g/L,PTM14.35 mL/L)的3 L發酵罐(美國NBS公司)中,初始攪拌轉速為500 r/min,通氣量為2 vvm,50%氨水及30%磷酸控制pH5.5,生長期培養溫度為30℃,當甘油耗盡溶氧反彈時以指數流加方式補加50%(w/v含 12 mL/L PTM2)甘油,待甘油再次耗盡溶氧反彈時,饑餓培養2 h,開始流加誘導培養基(100%甲醇含12 mL/L PTM2),同時溫度降低至22℃,攪拌轉速升高至900 r/min,誘導PGL表達。誘導培養基采用分階段流加方式:0~8 h流速2 mL/h,8~90 h流速 9.6 mL/h,>90 h流速 2 mL/h[20]。 每隔 12 h取樣一次,測定生物量、酶活、蛋白含量等參數。
1.2.5 指數流加指數流加是建立在對物料平衡及反應動力學兩方面進行合理假設基礎上的一種較為常見的前置流加控制法,可使限制性基質的供給與反應器中細胞量隨時間的指數增加相適應。流加速率為
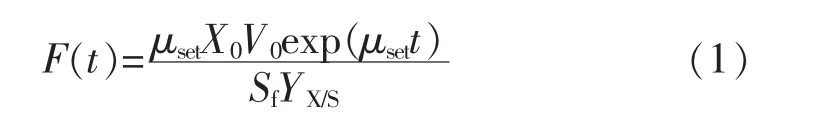
式中:F(t)為流加速率,L/h;X0為細胞密度,gcell/L;V0為初始體積,L;Sf代表補料液中甘油濃度,g/L;YX/S對底物細胞得率(gcell/gglycerol);μset為設定的比生長速率(h-1)。 其中 μset為 0.176 h-1,YX/S為 0.435 g/g,Sf為500 g/L[5]。
1.2.6 PGL蛋白純化取發酵液于4℃,8 000 r/min離心10 min獲得含PGL發酵上清,將發酵上清置于冰上,邊攪拌邊緩慢加入硫酸銨粉末,至終濃度40%~60%,離心得富集的粗PGL蛋白,粗蛋白用5 mL緩沖液A (20 mmol/L甘氨酸-NaOH溶液,pH 7.5)復溶,并置于A液中透析24 h,將樣品用0.22 μmol/L微孔濾膜過濾,純化使用AKTA純化儀及5 mL 陽離子柱(HiTrapTM SP FF 2.5 cm,GE)。柱純化條件如下:用5~10倍柱體積的緩沖液A平衡陽離子柱,之后以1 mL/min流速進樣,進樣結束以5個柱體積A液繼續平衡柱子,流速改為2 mL/min,用洗脫緩沖液B(20 mmol/L甘氨酸-NaOH溶液,pH 7.5,1 mol/L NaCl)梯度洗脫,收集洗脫液,取測得PGL酶活的洗脫液在A液中透析過夜,樣品4℃保存。
1.2.7 PGL活力測定酶活測定條件為:發酵液8 000 r/min離心10 min,胞外PGL即包含于發酵上清液之中,取一定量稀釋做檢測。PGL反應體系[21]:含0.2%聚半乳糖醛酸(底物)的甘氨酸-NaOH緩沖 液 (0.2 mol/L,0.44 mmol/L 的 CaCl2,pH 9.4)2 mL,待測樣品20 μL,無活性的酶液為空白對照。PGL反應條件為:將反應體系置于45℃下水浴15 min,用 3 mL 磷酸溶液(0.03 mol/L)終止反應,在235 nm處測定吸光度值。單位酶活定義:單位時間(1 min)裂解聚半乳糖醛酸產生1 μmol的不飽和聚半乳糖醛酸所用的酶量。計算方法如下

式中:b為比色皿厚度,cm;4 600為不飽和PGA于235 nm處的摩爾吸光系數,L/mol/cm;t為酶促反應時間(在酶促反應線性范圍之內),min。
1.2.8 熒光定量PCR目標基因拷貝數通過RTPCR 測定[18-19],定量 PCR 引物(表 1)。 20 μL反應體系包括 10 μL SYBR Premix Ex TaqII (Tli RNaseH Plus),0.8 μL RT-PCR F/R 引物 (10 μmol/L),2 μL DNA 模板(<100 ng),6.4 μL 滅菌蒸餾水。PCR 反應條件:95 ℃,30 s預變性;95 ℃,5 s,55 ℃,30 s,40個循環反應。40個循環后通過融解曲線分析特異性。以10倍梯度稀釋的內參基因GAPDH及目標基因作為模板建立標準曲線,以重組菌株基因組作為RT-PCR的模板,通過雙曲線法計算不同重組菌株中相關基因的拷貝數。
1.2.9 PGL去糖基化純化后的PGL用去糖基化酶 Endo H(美國 NEB 公司)處理[22]:10 μL PGL 上清,加入 1 μL 10×Glycoprotein Denaturing Buffer混合,100 ℃反應 10 min,再加入 2 μL 10×GlycoBuffer 3,2.5 μL Endo H,加水補足至 20 μL,37 ℃反應 1 h。
1.2.10 PGL半衰期的測定重組PGL及突變體的熱穩定性用55℃下酶活力半衰期(t1/2,min)來表示。將純化后的PGL用20 mmol/L甘氨酸-氫氧化鈉緩沖液(pH 7.5)稀釋至 100 μg/mL,并在 55 ℃保溫,間隔測定殘余酶活,將殘余酶活按照文獻所述方式擬合并計算t1/2[23]。
1.2.11 SDS-PAGE分析樣品與4×加樣Buffer混合,72℃處理10 min,5%濃縮膠及12%分離膠購自Invitrogen公司(美國),操作參考產品說明書。

圖1 密碼子優化后的PGL基因序列Fig.1 Optimized PGL gene sequence
2 結果與分析
2.1 密碼子優化對PGL表達的影響
P.pastoris對一些密碼子顯示出偏好性,從而影響編碼基因的表達效率[13]。研究發現,密碼子優化可使葡萄糖異構酶在P.pastoris中的表達效率提高2.4倍[24]。為提高PGL的轉錄及翻譯效率,基于P.pastoris的密碼子偏好性對Bacillussp.WSHB04-02來源的PGL基因進行優化(圖1),使其密碼子適應指數 (CAI) 由 0.63提高至 0.84[25],GC含量從46.64%降為42.41%。基因結構分析表明,不適宜的GC峰也被優化以延長mRNA的半衰期;影響mRNA穩定性及核糖體結合的莖環結構被破壞掉;消極的順式位點也被成功修飾。
將Bacillussp PGL基因克隆至pPIC9K中的EcoR I-NotI位點,構建得到PGL表達質粒pPIC9K-PGL (圖 2 (a)),轉化P.pastorisGS115(his-)得到重組菌GS115/PGL轉化子,經MD/MM平板篩選Mut+/Muts菌株,獲得表型為Mut+的數十株菌株,再經不同濃度梯度G418平板篩選,從1~4 mg/mL平板上各挑取10個單菌落進行搖瓶發酵96 h。其中,菌株GS115/PGL 9#、14#和19#胞外酶活分別為256.35、301.32 U/mL和273.24 U/mL。重組菌株PGL基因拷貝數使用RT-PCR進行確定(表2),PGL拷貝數為3時重組菌株具有最佳的PGL表達能力。未經密碼子優化的PGL基因克隆至pPIC9K中的EcoR I-NotI位點得到pPIC9K-PGLN,轉化P.pastorisGS115(his-)得到含3個拷貝數PGL基因的重組菌GS115/PGLN。在相同拷貝情況下,GS115/PGL 14#較GS115/PGLN胞外PGL酶活提高25.1%,表明密碼子優化能有效提高PGL在P.pastoris分泌表達。

圖2 重組質粒示意圖Fig.2 Schematic plot of recombinant plasmids

表2 重組菌生產性能及PGL基因拷貝數Table 2 Productivity of recombinant strains and copy numbers of PGL gene
2.2 共表達分子伴侶對PGL胞外表達的影響
共表達UPR相關分子伴侶可有效減緩未折疊蛋白效應,促進外源蛋白在P.pastoris中的表達[14-16]。ERO1可以輔助促進外源蛋白在內質網中的折疊,UBC1可以通過泛素化作用輔助錯誤折疊蛋白的降解,共表達ERO1及UBC1可有效促進外源蛋白正確折疊及錯誤折疊蛋白的快速降解,從而緩解細胞代謝壓力,促進外源蛋白高效表達。為提高PGL胞外表達水平,在重組菌GS115/PGL 14#中分別共表達ERO1及UBC1。將ERO1及UBC1基因克隆至pGAPZA中的Bsp119 I-KpnI位點,構建得到表達質粒 pGAPZA-ERO1(圖 2(b))和 pGAPZA-UBC1(圖 2(c)),電轉化 GS115/PGL 14#,得到 10 個轉化子進行搖瓶發酵。經過96 h甲醇誘導發酵,PGL酶活達到最大值,其中重組菌GS115/PGL-ERO1 6#及GS115/PGL-UBC1 3#的PGL酶活分別達到408.27 U/mL 及 368.54 U/mL(圖 3),較出發菌分別提高35.5%及22.3%。
為提進一步提高PGL產量,在重組菌GS115/PGL 14#中共表達 ERO1及 UBC1。將pGAPZAERO1用BglII與BamH I雙酶切,得到GAPERO1-AOX(TT)表達框,克隆至BamH I單酶切的pGAPZA-UBC1載體,得到共表達載體pGAPZERO1-UBC1(圖2d)。發酵結果顯示,GS115/PGLERO1-UBC1 2#重組菌株其最大酶活達到450.12 U/mL,相比于出發菌株提高49.3%(圖3)。

圖3 重組菌搖瓶發酵過程曲線Fig.3 Fermentation process curve of the recombinant strains in shake flask
以GAPDH基因為內參,應用RT-PCR測定了重組菌中分子伴侶的基因拷貝數。結果顯示,GS115/PGL-ERO1-UBC1 2#中ERO1與UBC1基因拷貝數分與GS115/PGL-REO1 6#和GS115/PGL-UBC1 3# 相同(表 3)。
2.3 3 L罐發酵培養
分批補料發酵是一種簡單有效的發酵模式,可在短時間內增加P.pastoris菌體濃度來提高目的產物的產量以及生產強度。通過分批補料發酵及相關參數優化,目標產物的產量可以提高數倍之多[26-27]。本實驗室前期研究PGL時,通過分批補料發酵策略提高了PGL的表達水平[5,7]。因此,比較了重組菌GS115/PGL 14#及GS115/PGL-ERO1-UBC1 2#在3 L罐中分批補料發酵。

表3 RT-PCR測定共表達基因拷貝數Table 3 Copy numbers of co-expressing gene detected by real-time PCR
發酵起始采取分批培養,在第19 h DO反彈初始甘油耗盡后,開始采用指數流加方式流加50%的甘油溶液,控制比生長速率(μset)為 0.176 h-1,指數流加19 h,當甘油耗盡DO再次反彈后,饑餓培養2 h后開始分階段 (0~8 h流速2 mL/h,8~90 h流速9.6 mL/h,>90 h流速2 mL/h)流加誘導培養基,誘導重組菌生產PGL[20]。如圖4所示,在20~40 h細胞生長較快,誘導發酵初期菌體依然緩慢生長,至發酵后期 (100~160 h)菌體量不再增加,此時重組菌GS115/PGL-ERO1-UBC1 2#的細胞干重可達120 g/L,組合共表達ERO1及UBC1的重組菌株GS115/PGL-ERO1-UBC1 2#在誘導96 h時PGL酶活達到最大值為1 362.31 U/mL,相比于未共表達分子伴侶的重組菌株GS115/PGL 14#的934.34 U/mL提高45.8%,且菌體生長更好。這些結果表明共表達分子伴侶ERO1及UBC1可有效促進菌體生長及外源蛋白表達。


圖4 重組菌3 L罐分批補料發酵過程曲線Fig.4 Fermentation process curve of the recombinant strains during fed-batch fermentation in the 3 L fermentor
為進一步研究糖基化對PGL表達的影響,通過陽離子柱純化方法從GS115/PGL-ERO1-UBC1 2#誘導96 h發酵上清中分離純化得到PGL純酶。圖5中SDS-PAGE分析顯示,純化后的PGL蛋白條大小為47 kDa和49kDa,較PGL理論分子量43.5 kDa偏大。純化的PGL經去糖基化酶Endo H處理后,PGL主要轉化為43.5 kDa蛋白及其它未知低分子量蛋白,上述結果表明,PGL在P.pastoris表達后發生了糖基化。研究表明,糖基化對酶分子在酵母表達水平有重要作用[22,28]。在下一步工作中,將通過去糖基化修飾來進一步提高PGL的胞外表達水平。此外,GS115/PGL-ERO1-UBC1 2#生產的PGL 55℃下酶活力半衰期較野生酶提高81.3%,達到1.16 h,穩定性明顯提升。
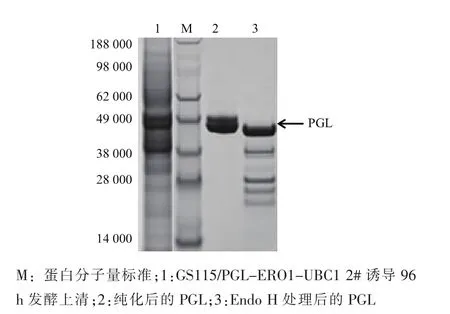
圖5SDS-PAGE電泳分析Fig.5 SDS-PAGE analysis
3 結語
本研究表明系統性的密碼子偏好性優化及共表達分子伴侶可顯著促進宿主細胞生長及外源蛋白PGL的表達。單獨共表達分子伴侶ERO1或UBC1對促進PGL的表達擁有較好效果,組合共表達ERO1-UBC1效果更加顯著,也說明兩個分子伴侶之間擁有積極的相互作用。在相同分批發酵條件下,改造后的重組菌GS115/PGL-ERO1-UBC1 2#胞外PGL酶活達到1 362.31 U/mL,較改造前PGL酶活 863 U/mL提高 57.9%[7]。此外,GS115/PGLERO1-UBC1 2#生產的PGL55℃下酶活力半衰期較野生酶提高81.3%,達到1.16 h,穩定性明顯提升。研究結果對促進PGL產量的提升及其應用具有重要現實意義。
