正構烷烴萘芳烴含量的測定
2019-04-11 07:39:08陳麗君黃愛忠
中國資源綜合利用 2019年3期
關鍵詞:分析
陳麗君,黃愛忠,秦 杰
(南京金陵石化烷基苯廠,南京 210046)
目前,南京金陵石化烷基苯廠測定正構烷烴產品中的芳烴含量時參照《液體石蠟中芳烴含量測定法(紫外分光光度法)》(SH/T0409-1992(2005))標準。該方法中,萘芳烴濃度測定取波長285 nm處吸光度A,代入計算公式A=abc,得出芳烴中萘的濃度c[1]。其中,吸收系數a為API研究項目的紫外光譜數據中各種萘化合物的平均吸收系數,平均吸收系數作為一種經驗值,直接用于計算不同正構烷烴產品萘芳烴的含量,存在一定誤差,試驗發(fā)現,其一般低于實際含量。為此,測定正構烷烴產品萘芳烴的含量,筆者使用ASTMD2008-12標準,方法中對萘芳烴含量的測定選定在波長290 nm處,測定不同濃度萘標樣的吸光度,然后據此做出標準曲線,得到吸收系數,在同樣條件下測量試樣的吸收光譜來確定其萘芳烴含量[2]。
1 試驗部分
1.1 儀器
島津紫外-可見分光光度計:UV-2600;石英比色皿:1 cm;容量瓶:25 mL,50 mL,100 mL;移液管1 mL;分析天平;分液漏斗50 mL;燒杯250 mL。
1.2 試劑和溶液
異辛烷:光譜純;萘:技術條件符合Q/CYDZ2457-2012,含量>=99.5%;濃硫酸:優(yōu)級純;無水碳酸鈉,含量99.95%~100.05%。
1.3 標準曲線的繪制
1.3.1 萘標準溶液的配制
稱100 mg的萘,在燒杯中完全溶解后,轉移至100 mL的容量瓶,加異辛烷稀釋至刻度,搖勻,配制成1 g/L的萘標準溶液。稱100 mg的萘,在燒杯中完全溶解后,轉移至25 mL的容量瓶,加異辛烷稀釋至刻度,搖勻,配制成4 g/L的萘標準溶液。
從配制好的1 g/L的萘標準溶液分別取5.00 mL、2.50 mL、2.00 mL、1.00 mL于50 mL容量瓶中,加異辛烷稀釋至刻度,搖勻。從配制好的4 g/L的萘標準溶液分別取1.00 mL于50 mL容量瓶中,加異辛烷稀釋至刻度,搖勻。
1.3.2 吸光度的測定
以異辛烷為參比溶液,在波長290 nm處,用1 cm比色皿在紫外-可見分光光度計UV-2600測定標準比色溶液的吸光度,結果如表1所示。以萘標液的濃度為橫坐標,從每個標準比色皿的吸光度中扣除試劑空白的吸光度為縱坐標,線性回歸:y=9.2765x+0.0088,相關系數r2=0.9993。萘標準曲線如圖1所示,萘標液的吸收光譜圖如圖2所示。

表1 萘標準溶液吸光度的測定

圖1 萘溶液標準曲線
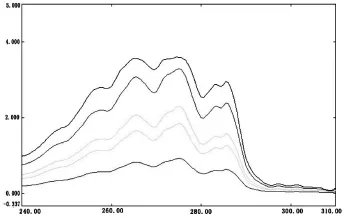
圖2 萘標液的吸收光譜圖
1.4 樣品測定
1.4.1 試液的配制
稱取5 g左右樣品,移至25 mL的容量瓶中,用異辛烷稀釋至刻度,搖勻。
1.4.2 測定過程
基線調零:用異辛烷充滿配對的吸收池,進行基線調零(220~400 nm),調整好基線后進行樣品分析;將待測量試液倒入1.0 cm比色皿,同時用異辛烷作參考,確保窗口干凈,將比色皿放入分光光度計的比色皿室并從220到400 nm波長開始掃描;掃描結束后,點擊“選點檢測”,獲取試樣在290 nm的吸光值A。
在指定的波長下計算樣品的吸收系數a,具體計算公式如下:

式中,A為在特定波長下的樣品溶液的吸光度減去比色皿的校正值;b為吸收池的光程,cm;c為吸收池中樣品的濃度,g/L;f為稀釋比例,即溶液體積與含有相同量溶質的原溶液的體積之比,對于初始溶液,f=1。
2 結果與討論
2.1 精密度的測定
對三份不同批次的試樣,按照上述方法,平行測定5次,結果如表2所示。由表2計算可知,其平均RSD為1.96%,該方法分析結果重復性、精密度高。
2.2 回收率測定
用一個已知萘芳烴含量的樣品,按上述分析過程,進行5次加標回收率試驗,結果如表3所示。由表3計算可知,其收率平均值為104.14%,回收率在100%~110%,說明該方法準確可靠,能滿足分析要求。
2.3 分析方法驗證
將用濃硫酸處理后除掉芳烴的輕蠟作為基液,配制成萘濃度為1 g/L、2 g/L、4 g/L的輕蠟樣。其中,樣品的密度為747.4 kg/m3。通過計算得到樣品實際萘含量,結果如表4所示。

表2 精密度試驗數據
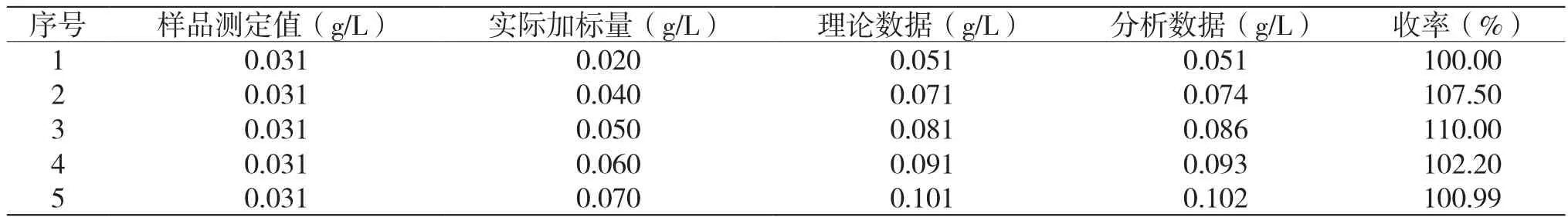
表3 回收率試驗數據
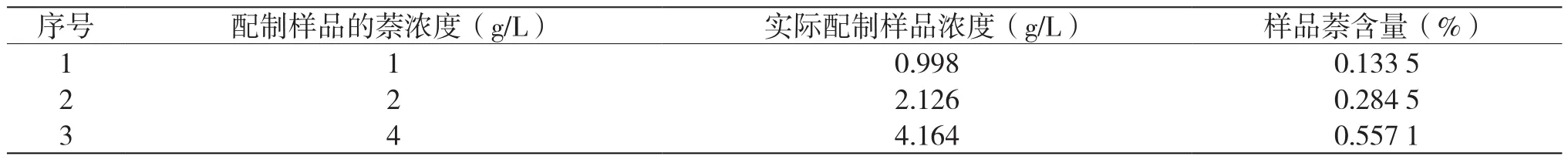
表4 樣品實際萘含量數據
將配制的已知萘含量的輕蠟樣,按上述分析過程,每一不同萘含量樣品分別平行測定3次。分析結果如表5、表6、表7所示。

表5 1 g/L的萘含量輕蠟樣分析結果

表6 2 g/L的萘含量輕蠟樣分析結果

表7 4 g/L的萘含量輕蠟樣分析結果
由表5、表6、表7可知,已知萘含量的輕蠟樣用ASTMD2008-2000分析方法測定的結果與實際理論值相符,說明此分析方法準確、可靠。
為了進一步驗證此方法,筆者選擇290 nm作為測定波長的準確性,選取了吸收光譜圖上受干擾最少的一段,進一步探究了287 nm、288 nm、289 nm、291 nm、292 nm作為測定波長的分析情況。首先用相同的方法分別繪制了5個波長的標準曲線,其中287 nm波長對應的曲線方程為:y=25.644x+0.0654,R2=0.9992;288 nm波長對應的曲線方程為:y=22.164x+0.0056,R2=0.9999;289 nm波長對應的曲線方程為:y=14.223x+0.0361,R2=0.9991;291 nm波長對應的曲線方程為:y=6.5029x+0.002,R2=0.9991;292 nm波長對應的曲線方程為:y=4.8786x-0.0032,R2=0.9995。筆者選用配制的1 g/L萘含量輕蠟樣,分別測定287 nm、288 nm、288 nm、291 nm、292 nm,作為測定波長的分析結果,分析結果如表8所示。
由表8可知,測定波長取290 nm的分析方法,對于測定萘芳烴含量更準確。
2.4 與原分析方法對比
測定8組不同批次的樣品,分別用方法SH/T0409-1992(2005)和方法 ASTMD2008-2000分析計算,分析結果如表9所示。
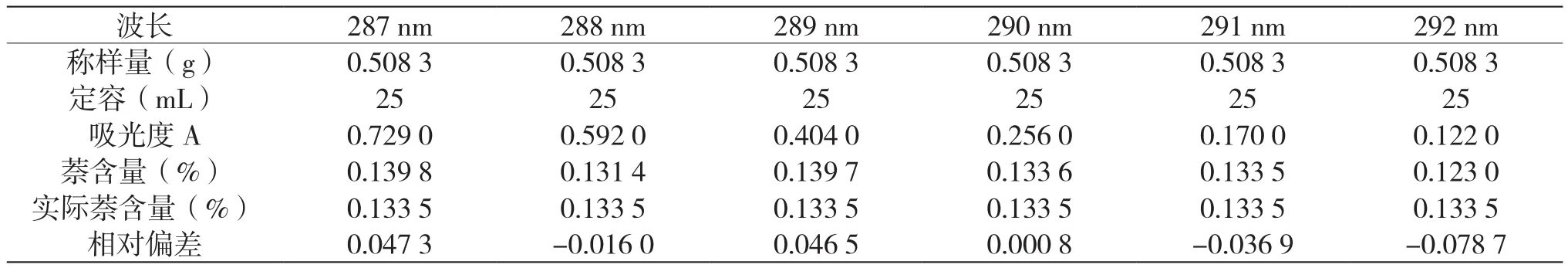
表8 1 g/L的萘含量輕蠟樣分析結果
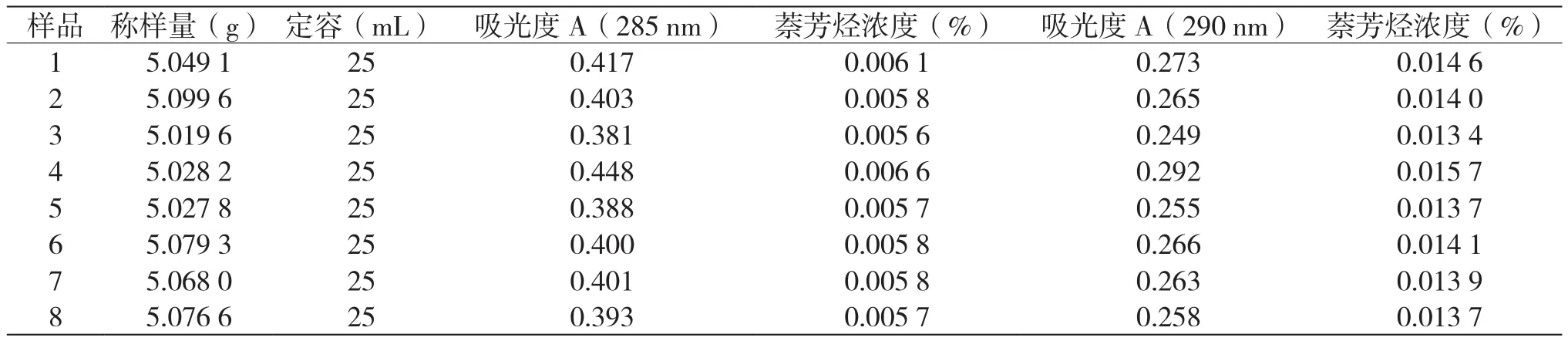
表9 8組不同批次樣品分析結果對比
由表9可知,方法SH/T0409-1992(2005)得到的分析結果明顯比ASTMD2008-2000分析結果偏低,SH/T0409-1992(2005)方法不適用于萘芳烴的測定。
3 結論
通過精密度試驗和回收率試驗,筆者初步判斷了此分析方法的準確性,由于分析方法中,測定萘芳烴的波長位置290 nm并不是最大吸收波長處,因此需要對290 nm波長的選擇做進一步驗證。這里將輕蠟通過濃硫酸處理,配制出已知萘濃度的輕蠟樣,濃度分別有1 g/L、2 g/L、4 g/L。針對不同濃度,筆者分別做了3組平行樣進行測定,試驗發(fā)現已知萘含量的輕蠟樣用ASTMD2008-2000分析方法測定結果與實際理論值相符,說明此分析方法準確、可靠。另外,1 g/L萘濃度輕蠟樣測定結果更接近理論值,配制過程引入誤差更小。于是,筆者選擇1 g/L萘濃度輕蠟樣進一步探究在受干擾最少的波長位置的不同波長的選擇對分析結果的測定影響,通過試驗探究進一步驗證了此分析方法選擇290 nm波長更具準確性。最后,比較了測萘芳烴的原方法SH/T0409-1992(2005)和此方法ASTMD2008-2000的分析結果,發(fā)現原方法得到的分析結果明顯比此方法的分析結果偏低,所以原方法不適用于萘芳烴的測定。
猜你喜歡
現代畜牧科技(2021年9期)2021-10-13 06:39:14
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業(yè)技術(2016年15期)2016-12-01 05:31:22
當代經濟研究(2016年5期)2016-12-01 03:12:05
現代農業(yè)(2016年5期)2016-02-28 18:42:46
出版與印刷(2016年3期)2016-02-02 01:20:11
中國中醫(yī)藥現代遠程教育(2014年11期)2014-08-08 13:23:44
華北水利水電大學學報(社會科學版)(2014年3期)2014-04-16 04:38:31
終身教育研究(2014年5期)2014-02-28 01:23:06
