三相萃取體系分離富集螺旋藻多糖及其結構特征分析
2019-04-04 08:37:58羅光宏馬明輝張喜峰楊生輝王丹霞陳天仁
食品與發酵工業 2019年6期
羅光宏,馬明輝,張喜峰*,楊生輝,王丹霞,陳天仁
1(河西學院,甘肅省微藻技術創新中心,甘肅 張掖,734000) 2(甘肅省河西走廊特色資源利用重點實驗室,甘肅 張掖,734000) 3(河西學院農業與生物技術學院,甘肅 張掖,734000)
螺旋藻(Spriulinaplatensis)是拉丁美洲和非洲國家的一種傳統食品。許多研究表明螺旋藻或其提取物能提高人體細胞免疫和體液免疫功能[1-2],對人類或其它動物具有抑癌作用[3-4]。從螺旋藻中提取的螺旋藻多糖,具有抗氧化、抗病毒、免疫活性、抗炎和DNA修復等多種生物學活性[5-6]。螺旋藻多糖可提高機體SOD的活力,提高酶促防御能力,消除自由基,改善活性氧產生的傷害;抗病毒作用機理通過阻斷病毒向宿主細胞吸附,抑制感染細胞內病毒復制過程;抗疲勞作用主要通過減少蛋白質和其他含氮化合物的分解代謝,降低血清尿素氮的形成,提高肝糖原和肌糖原貯備能力,明顯延長小鼠游泳運動耐力時間,起到抗疲勞作用;螺旋藻多糖構效關系主要體現在其一級結構和其生物學活性,如螺旋藻多糖相對分子質量、硫酸化等化學修飾、支鏈分支度結構改變,均對其生物學活性產生影響;如相對分子質量為1 000萬螺旋藻多糖比臨床多糖制劑激活體外單核細胞能力較強;螺旋藻多糖經硫酸化修飾后,具有更強抑制血管內皮細胞增殖獲活性等[7];螺旋藻多糖是一種水溶性多糖,可通過熱水浸提、沉淀、去蛋白和各種sephadex、sephacryl和sepharose柱層析方法等一系列步驟進行純化[8]。但是上述分離方法具有處理量大、得率較低、大量有機溶劑消耗和成本較高等缺點或局限性。此外,TCA(tricarboxylic acid cycle)法、酶解法和Sevage法包括化學試劑和復雜純化過程,已成為多糖中去除蛋白質的普遍方法[9-10]。
與傳統方法相比,三相萃取技術是一種新興的簡單、快速、高效、成本較低,非色譜分離技術[11]。三相萃取通常由一定量的無機鹽(如(NH4)2SO4)、含有蛋白質的粗提取液和一定量的有機溶劑(如叔丁醇)混合組成[12]。色素、脂類、抑制劑和疏水化合物富集在上相即叔丁醇相,中間相主要為富集的蛋白質或酶,而糖和其他親水性物質都集中在下層水相即硫酸銨相。上述三相的形成主要與鹽的離子強度、kosmotropy、表面張力增強、滲透應力有關[13]。由于其溫和液相環境在分離和純化生物活性物質方面具有舉足輕重作用。近年來,三相萃取已廣泛應用于蛋白質、酶、抗氧化物、油脂、金屬回收等成分的分離和純化[14-17]。
本試驗采用三相萃取技術分離純化螺旋藻多糖,分別研究(NH4)2SO4濃度、叔丁醇加入量、pH、萃取溫度、時間對螺旋藻多糖得率的影響,并對其結構特征進行了分析,以期為螺旋藻多糖深入研究奠定基礎。
1 材料與方法
1.1 材料、試劑與儀器
螺旋藻,由甘肅省微藻創新技術中心提供;(NH4)2SO4、叔丁醇、苯酚、濃H2SO4、葡萄糖,均為分析純;透析袋(分子截留量8~12 kDa),成都德思特生物技術有限公司。
PL-203 電子天平,梅特勒-托利多儀器有限公司;紫外可見分光光度計,上海譜元儀器有限公司;TG/DTA系列熱重差熱綜合熱分析儀,寧波賽茵儀器有限公司;氣相色譜質譜聯用儀,江蘇天瑞儀器股份有限公司;高溫凝膠滲透色譜儀,北京億路達機電設備有限公司;FEI Quanta 450 FEG場發射掃描電鏡,FEI公司。
1.2 實驗方法
1.2.1 螺旋藻多糖粗提液制備
準確稱取10 g螺旋藻粉末,按照固液比1∶0.045(g∶L)加入蒸餾水,在90 ℃水浴處理120 min后,5 000 r/min離心10 min后,上清液為螺旋藻多糖的粗提液[4]。
1.2.2 三相萃取螺旋藻多糖
取1.2.1制備的螺旋藻多糖粗提液10 mL,加入一定量的(NH4)2SO4使其終質量濃度為100~600 g/L,采用HCl或NaOH調節其體系pH為5~9,再加入一定體積的叔丁醇(5~30 mL),充分混勻后,在20~40 ℃ 水浴處理15~90 min,然后在3 000 r/min離心處理10 min,叔丁醇相可通過減壓蒸發回收叔丁醇;收集含多糖和硫酸銨的下相,記錄其體積,采用蒸餾水透析除鹽后,濃縮干燥得到螺旋藻多糖。
1.2.3 多糖得率測定及計算
多糖含量的測定采用苯酚-硫酸法,以100 μg/mL葡萄糖溶液為標準品,所得標準曲線的回歸方程為:y=0.113x+0.011 5(R2=0. 996 4)。其中y為490 nm 處的吸光度,x為葡萄糖的質量濃度(μg/mL)。
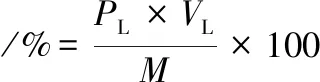
(1)
其中:CL和VL分別為三相萃取平衡后下相中螺旋藻多糖質量濃度和體積;M為螺旋藻粉末質量。
1.2.4 多糖含量測定及計算
將1.2.2制備的多糖加入一定體積蒸餾水,充分混勻后,測定多糖濃度,按式(2)計算多糖含量。
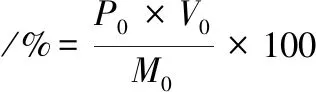
(2)
其中:P0和V0分別為溶解后多糖溶液質量濃度和體積,M0為制備的螺旋藻多糖。
1.2.5 與傳統多糖純化方法的比較
1.2.5.1 堿液冷提法
參考李會端和秦志玉之法[18],按照料液比1∶0.025(g∶L)比例加入濃度為0.4 mol/L NaOH,浸提2 h后,在4 000 r/min離心10 min,取上清液加入5% 的三氯乙酸溶液脫蛋白,采用95%醇沉后,按照1.2.2和1.2.3方法測定多糖得率和含量。
參考賁永光等的方法[19],按照固液比1∶0.025 (g∶L),在溫度為50 ℃,超聲功率320 W,提取50 min條件下,離心后取上清液,濃縮后醇沉,并采用丙酮洗滌后,按照1.2.2和1.2.3方法測定多糖得率和含量。
1.2.5.3 傳統回流提取法
參照張文雄等的方法[20],以固液比1∶0.015(g∶L),在溫度為60 ℃,超聲功率320 W,提取4 h,醇沉,透析處理后,按照1.2.2和1.2.3方法測定多糖得率和含量。
1.2.5.4 酶解結合超聲波提取法
參照王以斌等的方法[21],以固液比1∶0.016(g∶L),在溫度為60 ℃,超聲功率200 W,加入復合酶,提取時間15 min,經離心醇沉后,按照1.2.2和1.2.3方法測定多糖得率和含量。
1.2.6 螺旋藻多糖理化性質
1.2.6.1 螺旋藻多糖紫外-可見波長掃描
本實驗是微組織(微載體復合脂肪干細胞的3D培養)對周圍神經再生的體外研究。利用SD乳鼠脂肪干細胞分離培養后與多孔明膠微載體材料復合構建微組織,培養3 d后的微組織與背根神經節共培養,觀察其對背根神經節軸突生長的促進作用,并與2D細胞共培養以及單純背根神經節比較,并從細胞增殖和凋亡等方面分析比較2D、3D細胞培養優劣勢。
配制1 mg/mL螺旋藻多糖溶液,在190~400 nm進行掃描,檢測其是否含有蛋白質和核酸。
1.2.6.2 螺旋藻多糖紅外光譜檢測
將冷凍干燥后螺旋藻多糖進行溴化鉀壓片處理,在4 000~500 cm-1進行紅外光譜掃描,分析其官能團組成。
1.2.6.3 螺旋藻單糖組成檢測
參照范嘉龍等的方法[22],對螺旋藻單糖進行酸水解及衍生化處理后,采用氣相色譜-質譜法(GC-MS)分析其單糖組成及比例。
1.2.6.4 螺旋藻多糖分子質量測定
采用體積排阻色譜-光散射聯用(size-exclusion chromatography combined with laser light scattering,SEC-LLS)進行測定。多角度激光光散射儀λ=690 nm,校準常數=8.850 0e-6;色譜柱型號為7.8 mm×300 mm; 溶劑為水。
1.2.6.5 螺旋藻多糖掃描電鏡觀察
采用Quanta 450場發射掃描電子顯微鏡對冷凍干燥后多糖樣品噴金處理后,觀察其表面結構。
1.3 數據統計及處理
以Orgin 8.0作圖,所用實驗均重復3次,采用Mean±SD表示。
2 結果與討論
2.1 三相法分離富集螺旋藻多糖
2.1.1 硫酸銨質量分數對螺旋藻多糖得率的影響


圖1 不同影響因素(硫酸銨質量分數、叔丁醇加入量、pH、萃取溫度、萃取時間)對螺旋藻多糖得率的影響Fig.1 Effect of mass fraction of (NH4)2SO4, amount of t-butanol, pH, temperature, and extraction time on the yield of PSP
2.1.2 叔丁醇加入量對螺旋藻多糖得率的影響
與其他醇類溶劑相比,叔丁醇是一種獨特一元醇,除具有溶解非極性成分外,還有醇沉和浮選劑作用,因此,叔丁醇的加入量對螺旋藻多糖得率的影響如圖1-b所示。隨著叔丁醇加入量逐漸增加,當叔丁醇加入量為15 mL時,螺旋藻多糖得率為8.52%。原因可能是隨著叔丁醇加入量增加和硫酸銨質量分數的逐漸減小所呈現協同效應增強,表明叔丁醇作為一種kosmotrope試劑,對硫酸根增強效應具有重要作用,少量叔丁醇對硫酸銨協同效應較弱[24];大量叔丁醇加入會導致下相中多糖含量減少,促使叔丁醇和硫酸根共同競爭水分子,導致得率下降。因此,確定叔丁醇加入量為15 mL。
2.1.3 體系pH對螺旋藻多糖得率的影響
固定硫酸銨質量分數為20%,采用HCl或NaOH調節體系pH分別為5、6、7、8、9,加入叔丁醇15 mL,在25 ℃水浴60 min后,收集下相體積,計算多糖得率。結果如圖1-c所示。當體系pH為7時,有利于螺旋藻多糖從三相中分離和富集,多糖得率最高為8.79%;原因可能pH影響生物大分子如蛋白質解離或其表面電荷。此結果與劉楊[25]的研究結論是一致的,因此,確定三相體系pH為7。
2.1.4 萃取溫度對螺旋藻多糖得率的影響
固定硫酸銨的質量分數為20%,調節體系pH為7,加入15 mL叔丁醇,分別在20、25、30、35、40 ℃水浴處理60 min后,收集下相體積,計算多糖得率。結果如圖1-d所示,在20~35 ℃,多糖得率逐漸升高,此結論與DENNISON等[26]研究結果一致,即溫度升高后,有利于多糖分子羥基基團的暴露,利于氫鍵網絡的形成,并促使其親水性增強,在下相中多糖含量增加,得率升高;當溫度超過35 ℃后,高溫所需能耗增加。因此,本試驗確定萃取溫度為35 ℃。
2.1.5 萃取時間對螺旋藻多糖得率的影響
萃取時間不僅影響目標產物質量和得率,且影響整個生產成本。因此,研究萃取時間對螺旋藻多糖得率的影響。由圖1-e可知,萃取時間對螺旋藻多糖得率影響不顯著,當萃取時間為30 min后,萃取時間繼續增加,螺旋藻多糖得率變化無顯著差異。因此,確定萃取時間為30 min。
2.2 三相萃取螺旋藻多糖與傳統提取方法的比較
不同方法提取螺旋藻多糖得率和純度如表1所示。

表1 不同方法提取螺旋藻多糖得率和純度的比較研究Table 1 Comparison of yield and purity of polysaccharide from Spirulina platensis by different methods
由表1可知,采用三相萃取螺旋藻多糖方法與傳統方法提取相比,該法制備的多糖具有較高的得率和含量,具有分離和富集多糖的作用;采用三相萃取后,目標產物多糖主要分布在下相,蛋白質主要富集在中間相形成固體,上相主要含有非極性物質,可有效改善多糖純度,因此,三相萃取作為一種有效的萃取分離方法,具有同時分離和富集目標產物的作用。
2.3 螺旋藻多糖理化性質
2.3.1 多糖紫外掃描圖譜
由圖2可知,在190~400 nm,260和280 nm處無吸收峰,表明該多糖溶液中不含核酸和蛋白質。

圖2 螺旋藻多糖紫外可見波長掃描Fig.2 UV-vis wavelength scanning of the purified PSP
2.3.2 紅外光譜分析
由圖3可知,在4 000~500 cm-1,在3 384.81 cm-1和2 928.81 cm-1處具有O—H和C—H伸縮振動峰。

圖3 純化后螺旋藻多糖紅外光譜Fig.3 The FT-infrared spectra of the purified PSP
1 652.76 cm-1和1 373.74 cm-1處具有酯化羰基和羧基的伸縮振動峰。1 080.67 cm-1和1 035.14 cm-1處有吸收峰,說明螺旋藻多糖為吡喃糖。931.66 cm-1和762.03 cm-1有吸收峰,說明吡喃環有2種差向異構體。
2.3.3 螺旋藻多糖的單糖組成
在相同處理條件下,保留時間是樣品單糖組成定性分析依據之一,采用GC對6種單糖標準品進行測定,由圖4-A可知,相應色譜峰保留時間分別為:1. 鼠李糖(保留時間tR=12.95 min); 2. 來蘇糖(tR=13.57 min); 3.木 糖(tR=14.06 min); 4. 甘露糖(tR=19.65 min); 5.葡萄糖(tR=20.20 min); 6. 半乳糖(tR=21.20 min);對螺旋藻多糖水解單糖組成和標準單糖對比,由圖4-B可知,螺旋藻多糖主要由鼠李糖、甘露糖和葡萄糖組成,其單糖組成物質的量比值為5.14∶5.78∶81.37。

圖4 標準單糖(A)和螺旋藻多糖(B)的GC圖Fig.4 GC spectrum of monosaccharide references (A) and PSP (B)
2.3.4 螺旋藻多糖分子質量測定
由圖5可知,螺旋藻多糖重均分子質量Mw為3.597×104, 數均分子質量為Mn=2.189×104,Mw/Mn=1.643,表明分子質量分布具有均勻性。
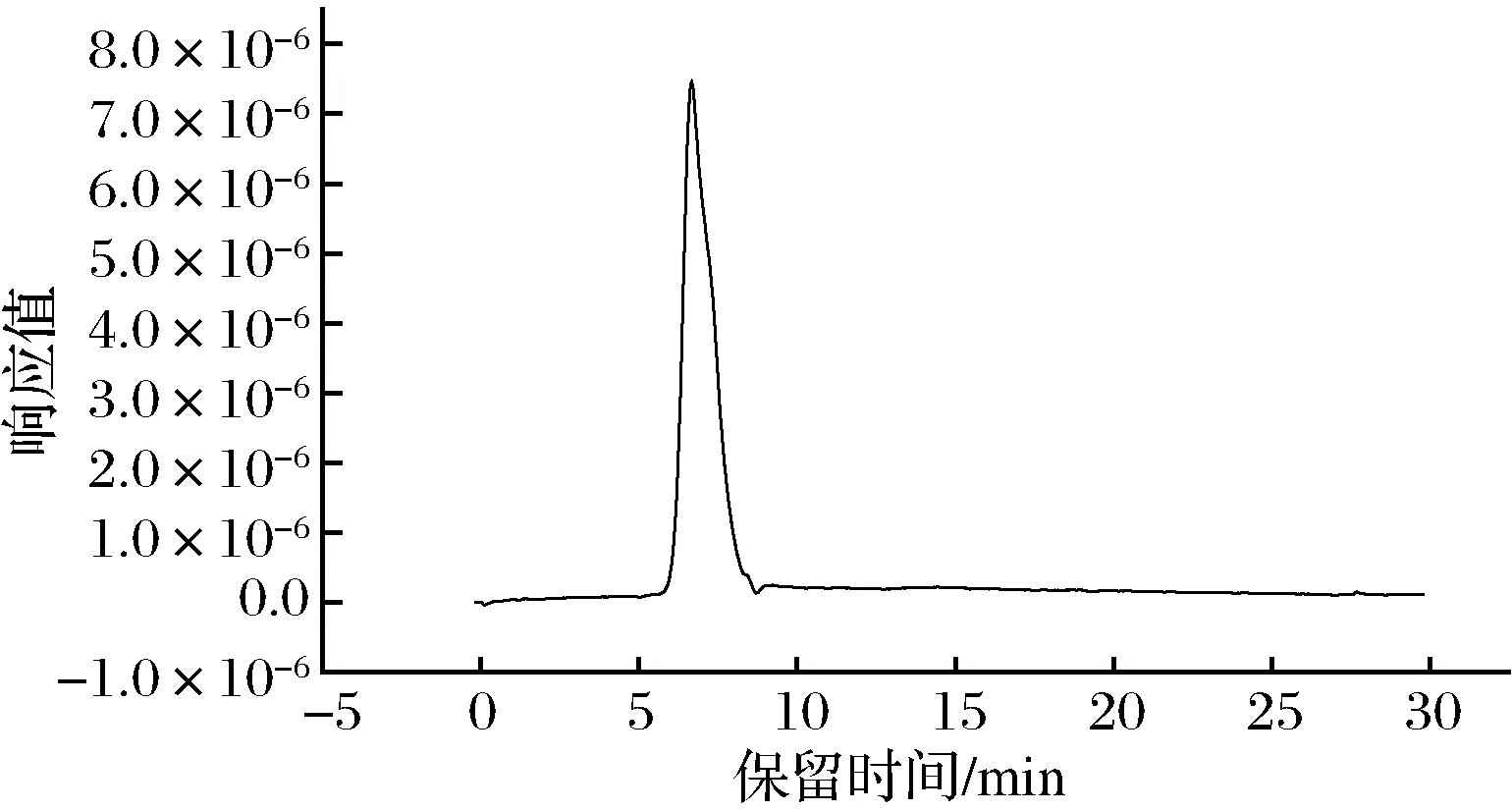
圖5 螺旋藻多糖SEC-LLS結果Fig.5 SEC-LLS of PSP
2.3.5 螺旋藻多糖掃描電鏡形態觀察
圖6為不同放大倍數下三相萃取后螺旋藻多糖掃描電鏡圖,在5 000×放大倍數下觀察,螺旋藻多糖表現為具有致密結構的球狀;10 000×放大倍數下,其呈現均一緊密球狀結構;由于掃描電鏡技術用來觀察物質表面狀態,其結果可能與樣品干燥方法、外力作用等因素有關;有關其多糖內部結構及其連接方式,有待進一步分析。

A-10 000×;B-5 000×圖6 螺旋藻多糖掃描電鏡圖Fig.6 Scanning electron microscope(SEM) photographs of the PSP
3 結論
本試驗采用三相萃取技術對螺旋藻多糖的分離和富集進行了初步研究,并對制備的多糖結構特征進行了分析,為深入研究多糖結構與生物活性之間的關系奠定基礎。
在三相萃取過程中,硫酸銨質量分數、叔丁醇加入量、體系pH、萃取溫度和時間均對多糖得率產生一定的影響;通過紫外光譜、紅外光譜、掃描電鏡、凝膠滲透色譜、單糖組成檢測對三相萃取制備的螺旋藻多糖結構進行了表征。當硫酸銨質量分數為20%,叔丁醇加入量為15 mL,體系pH為7.0,在35 ℃條件下,萃取30 min后,螺旋藻多糖得率為(9.25±0.42)%, 含量為(86.17±0.10)%。分子質量測定表明其重均分子質量為3.597×104;螺旋藻多糖主要由3種單糖組成,其摩爾比為:n(鼠李糖)∶n(甘露糖):n(葡萄糖)=5.14∶5.78∶81.37;掃描電鏡結果顯示其為結構緊密的球狀,在一定程度上反應其與樣品處理方式有關。
與傳統的多糖提取方法相比,三相萃取屬于一種綠色、具有廣闊前途的萃取技術,具有同時分離目標產物和除去其他雜質的特性,可為其他天然產物中活性成分的有效分離提供技術支撐。
猜你喜歡
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
兒童故事畫報(2019年5期)2019-05-26 14:26:14
產品可靠性報告(2017年7期)2017-09-05 09:49:12
Coco薇(2016年2期)2016-03-22 02:42:52
汽車觀察(2016年3期)2016-02-28 13:16:26
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
