氣相色譜-質譜法測定組合工藝己內酰胺產品中的微量關鍵雜質
2019-04-02 12:10:12韓江華
色譜 2019年4期
關鍵詞:工藝
韓江華
(中國石油化工股份有限公司石油化工科學研究院, 北京 100083)
己內酰胺(caprolactam, CPL)是生產聚酰胺合成纖維的重要原料,工業生產有多種技術路線,但主要生產方法是基于環己酮肟的Beckmann重排反應和經由六氫苯甲酸的甲苯法路線[1]。近年又開發出了將上述兩種方法結合起來生產CPL的組合工藝法技術路線,可以在副產相同產量硫酸銨的情況下多產CPL,這是一種具有世界領先水平的先進技術。
CPL的純度及其他質量指標對聚合產品的質量有重要影響。研究[2]表明Beckmann重排產物中的某些雜質能與CPL結構中的胺基、酰基反應生產低分子聚合物,影響聚合產物質量。CPL中殘留環己酮肟、環己酮、苯胺等的含量增大時,聚酰胺的黏度將降低。特別是產品中含有硝基苯類雜質時將使聚合物的顏色加深,影響產品外觀。其他雜質如醇類、甲基CPL、八氫吩嗪等并不影響聚合物的相對分子質量,但八氫吩嗪會影響到聚合產物的顏色,影響最終產品的后續著色。
存在于CPL中的雜質與其生產路線密切相關,主要來源于原料中的雜質、多步制備過程中的各種副反應等。一般情況下CPL的產品質量主要由一些關鍵雜質的含量確定,但其評價手段則主要采用傳統而又簡單的GOST 7850-74(USSR)[3]或者ISO/TC47/15 (Netherlands)[4]標準法。這些簡單方法主要控制5項指標:(1)高錳酸鉀指數,主要測定其中過氧化物、環己醇、環己酮肟、苯胺、環己酮、環己酰亞胺、氨基己酸、八氫吩嗪等具有還原性的物質對高錳酸鉀的綜合作用;(2)揮發性堿,主要確定鏈狀脂肪酰胺、脂肪胺、芳香胺等在強堿性和近100 ℃條件下能夠通過揮發釋放出堿性物質的雜質含量;(3)紫外吸收值,測定290 nm的吸收值,確定產品中苯胺、硝基苯、八氫吩嗪、甲基苯胺、環己酰亞胺等帶有生色基團的雜質對光密度的影響;(4)色度;(5)酸堿性。這5項指標被稱為CPL的產品質量指標,優級品、一級品、二級品等都有相應的數值。這些數值盡管含有一定的物理意義,但不能提供具體的雜質信息。CPL的生產方法較多,其中的雜質也各不相同,每種雜質究竟在何種程度上影響上述測試數據,目前尚不清楚。但這些參數的變化對聚合物性能的影響已有結論[5],即高錳酸鉀數值的降低將降低聚合速度,產生不規整聚合物,并伴隨明顯的黏度降低;揮發性堿值較高的CPL制得的是相對分子質量較低的聚合物;高紫外吸收值對應的是有色的聚合物。
六氫苯甲酸-環己酮肟聯產CPL組合工藝涉及環己酮肟法和甲苯法兩種主要工藝,因此由組合工藝生產的CPL成品中的雜質有可能包括兩種工藝中都涉及的雜質,以及環己酮肟法和甲苯法中都不涉及的新生雜質。該技術在工業試生產時出現最終產品光密度值嚴重超標的問題,本工作采用GC-FID和GC-MS技術對其光密度不合格產品進行檢測,通過GC-MS技術確定了雜質的結構,并對雜質的來源進行了推測。
1 實驗內容
1.1 儀器、試劑及樣品
Agilent 6890N氣相色譜儀配Agilent 5972質譜儀、05版NIST庫(美國Agilent公司)。無水乙醇和二氯甲烷均為分析純,購自北京精細化學品公司產品。樣品取自石家莊化纖廠CPL生產線成品出口,用去離子水或者無水乙醇、二氯甲烷配成質量分數為50%的溶液,置于色譜樣品瓶中密封待用。去離子水為實驗室自制。
1.2 分析條件
氣相色譜-質譜分析條件:色譜柱為HP-INNOWAX, 60 m×0.25 mm×0.25 μm(美國Agilent公司)。柱溫:初始溫度為80 ℃,升溫速率為10 ℃/min,終溫為230 ℃,保持30 min。色譜柱流速為1 mL/min。分流進樣,分流比為10∶1。進樣量為0.5 μL。進樣口溫度為230 ℃。載氣為高純He,恒流。
質譜條件:電子轟擊離子源;離子阱溫度為150 ℃;傳輸線溫度為230 ℃;掃描范圍為35~450 u;轟擊電壓為70 eV。
氣相色譜分析條件:色譜柱為HP-FFAP, 30 m×530 μm×1 μm(美國Agilent公司)。柱溫為100 ℃, 以4 ℃/min的速率升溫至220 ℃,穩定30 min。色譜柱流速為2.5 mL/min;分流進樣,分流比為10∶1;進樣量為1 μL;進樣口溫度為230 ℃;載氣為高純N2,恒壓;檢測器溫度為250 ℃;尾吹流速為20 mL/min。
2 結果與討論
2.1 樣品性質
組合工藝生產的CPL由甲苯法和氨肟化得到的環己酮肟經Beckmann重排反應后制得,其中環己酮肟重排所用的催化劑即是由甲苯法得到的酰胺化液中所含的發煙硫酸,可見采用組合工藝可以提高CPL產能而不增加硫酸銨的產量。
顯然組合工藝生產的CPL中的雜質主要由甲苯法的雜質、Beckmann重排反應雜質和組合后新生產的雜質3部分構成。對甲苯法生產的CPL而言,反應結束后經過后續的中和、蒸餾、堿蒸餾和精餾等提純工藝可以獲得純度為99.98%的CPL成品。但組合工藝工業試驗時采用與甲苯法同樣的精制流程得到的產品光密度超出合格指標20多倍,最高時可達30倍,產品的其他指標如揮發性堿、高錳酸鉀值等都可達優級品。結果表明,產品中基本不含還原性物質,在100 ℃氫氧化鈉水溶液中不發生水解并產生揮發性的堿性物質(氨氣或者有機胺),組合工藝中產生的雜質的物理性質與CPL相近,很難通過目前的精制手段與CPL分離。
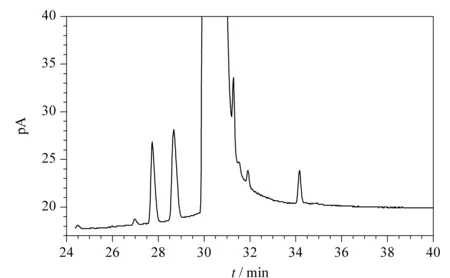
圖 1 組合工藝得到的己內酰胺(CPL)的色譜圖Fig. 1 Chromatogram of caprolactam (CPL) from the combined process The UV value of the product was 0.887 and the peak at a retention time of 34.149 min corresponded to the novel impurity.

圖 3 組合工藝得到的CPL的總離子流圖Fig. 3 Total ion chromatogram of CPL from the combined process The UV value of the product was 1.053 and the peak at a retention time of 18.30 min corresponded to the novel impurity.
2.2 樣品的GC-FID和GC-MS分析
甲苯法和氨肟化法得到的CPL中的雜質分析主要采用GC技術,色譜柱為極性較強的HP-FFAP或者HP-INNOWAX毛細管柱。兩種方法中的雜質有重疊也有不同,特點是甲苯法產物中雜質較多,分布較寬,雜質中除了有CPL的同系物甲基CPL外,還有甲苯法工藝產物中特有的雜質六氫苯甲酰胺等,隨雜質性質不同分布于色譜圖上主峰的兩側。而氨肟化法產物中的雜質主要是CPL同系物和原料環己酮肟,雜質數量少且出峰位置位于CPL主峰之后。因此,對組合工藝得到的CPL在與甲苯法和氨肟化法得到的CPL在同樣的條件下進行GC法分析,即可判斷出新雜質(見圖1和圖2)。GC-FID分析表明,在六氫苯甲酰胺色譜峰附近出現了一個新的色譜峰。樣品中加六氫苯甲酰胺純品重復試驗,新的色譜峰與六氫苯甲酰胺的色譜峰不重疊,表明是新雜質。色譜圖中的其他雜質都是甲苯法和氨肟化Beckmann重排法得到的CPL中常見的雜質,對產品質量指標影響不大。
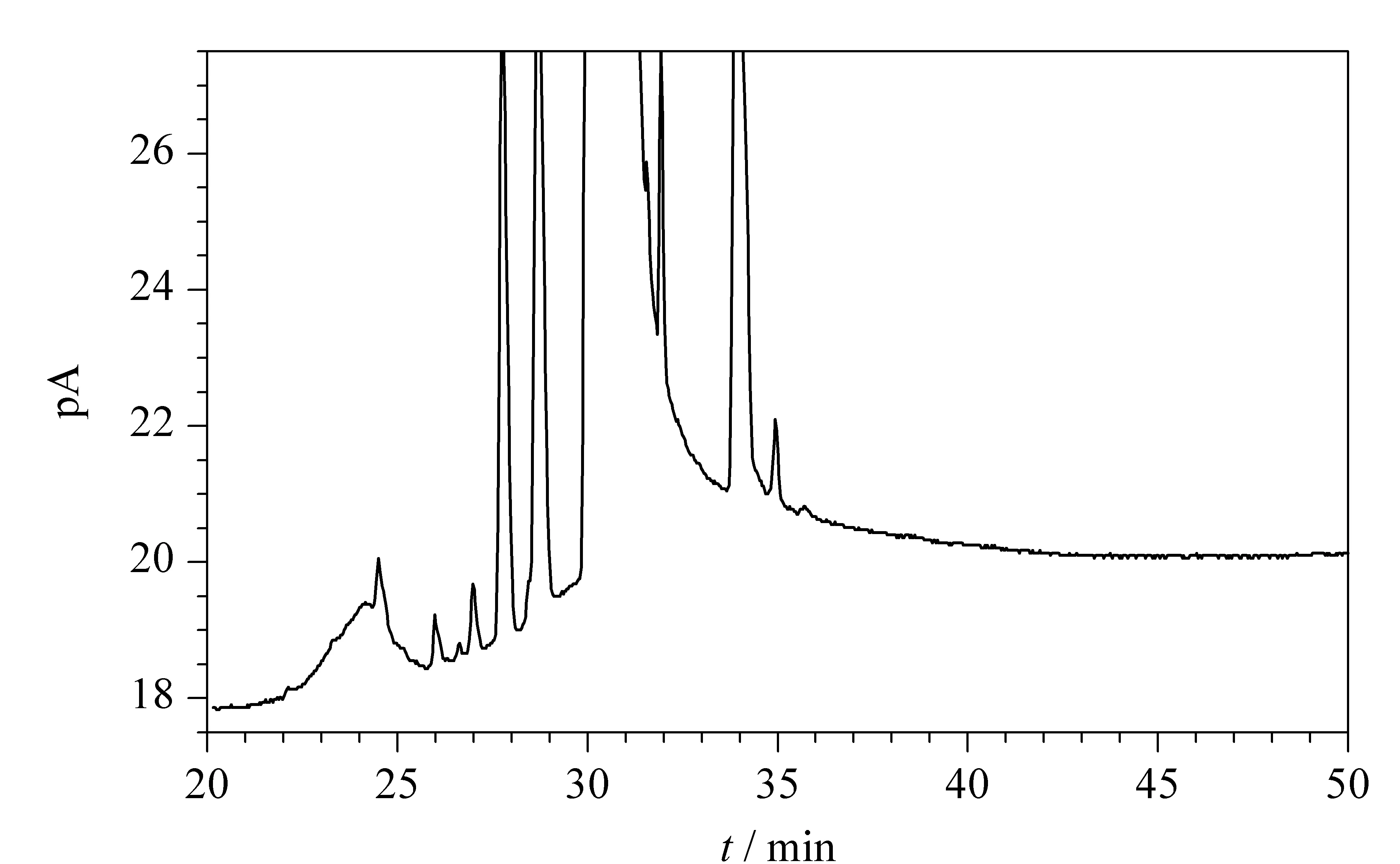
圖 2 組合工藝得到的CPL加六氫苯甲酰胺后的色譜圖Fig. 2 Chromatogram of CPL from the combined process containing hexahydrobenzamide The UV value of the product was 0.887 and the peaks at retention times of 33.957 and 34.926 min corresponded to the added hexahydrobenzamide and the novel impurity, respectively.
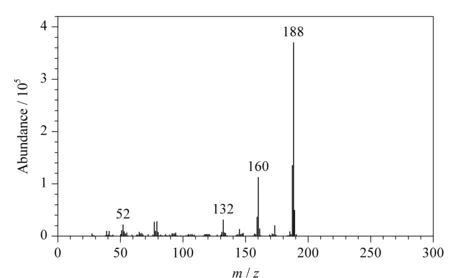
圖 4 組合工藝得到的CPL中新雜質的質譜圖Fig. 4 Mass spectrum of the novel impurity in CPL produced from the combined processThe UV value of the product was 1.053.
采用GC-MS技術確定雜質相對分子質量為188,總離子流圖以及質譜圖見圖3和圖4。早期文獻[6-14]表明環己酮肟Beckmann重排反應的關鍵副產物是八氫吩嗪,相對分子質量為188,但質譜分析儀所帶NIST庫中沒有這個化合物的質譜碎片信息,市場上沒有八氫吩嗪試劑出售,定性難度較大,質譜碎片解析是必須依靠的技術手段。質譜分析結果表明,八氫吩嗪是被高度懷疑的雜質。雜質的相對分子質量為偶數,根據氮數規則[15],如果不含氮原子并且不計氫的同位素貢獻,則根據M+1的豐度13.91可以計算出雜質分子所含碳原子數約為12,如果雜質分子中含有偶數氮原子且暫定氮原子數為2時,氮對M+1的貢獻值為0.74,此時如果忽略氫的貢獻,碳原子數仍為12,因此暫定分子中含有12個碳原子。碎片中有M-27的碎片離子m/z161,這是分子離子失去HCN的信息,表明雜質分子中含有氮原子,結合偶數分子量確定含有兩個氮原子,12個碳原子對M+2峰的豐度貢獻值為0.87,與實測值0.82相近而且已經超過實測值,表明雜質分子中可能不含氧原子。這樣確定雜質的分子式為C12H16N2,其環加雙鍵數為6,結構與八氫吩嗪一致。再結合組合工藝涉及的反應物以及組合工藝反應過程本身特點等推測,雜質就是八氫吩嗪。其典型的碎片離子及原始豐度和重新歸一化后的豐度數據見表1。主要碎片離子m/z是52、53、77、78、79、80、120、132、145、159、160、173、187、188和189。這些數據在非標準調諧狀態下獲得,計算值與實測值之間的誤差可能即由此引起。
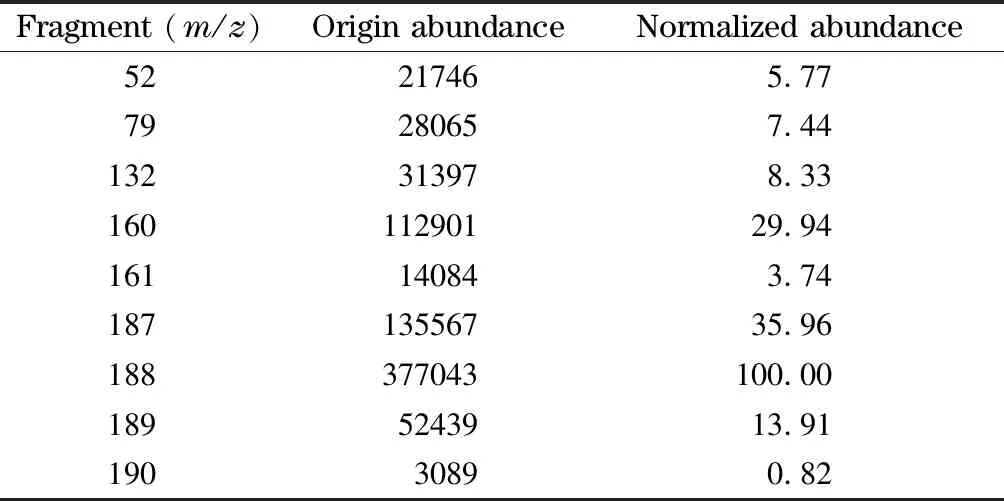
表 1 組合工藝得到的CPL新雜質的質譜碎片離子及其豐度Table 1 Main mass spectrum fragments and their abun- dances for the novel impurity in the CPL produced from the combined process
2.3 雜質的質譜碎片
八氫吩嗪結構中主要是近似于吡啶結構的碳氮雙鍵、近似于苯環或者環己烯結構中的碳碳雙鍵和環己烷中的碳碳單鍵,其電離能依次為9.3、9.2、8.8和9.9 eV,可見在電子轟擊條件下首先發生電離的是環己烯中的雙鍵。因此有如下的共振結構式,并在此基礎上得到分子離子峰m/z188,然后分子離子以逆Diels-Alder反應[15,16]方式裂解得到m/z為134、160、54、132、106等一系列碎片離子(見圖5)。m/z134碎片進一步進行逆Diels-Alder反應方式裂解即得m/z為79的吡嗪,吡嗪脫去氫氰酸即得m/z52碎片,其中m/z94碎片可看作雙游離基正離子。這樣可解釋大部分主要碎片離子。
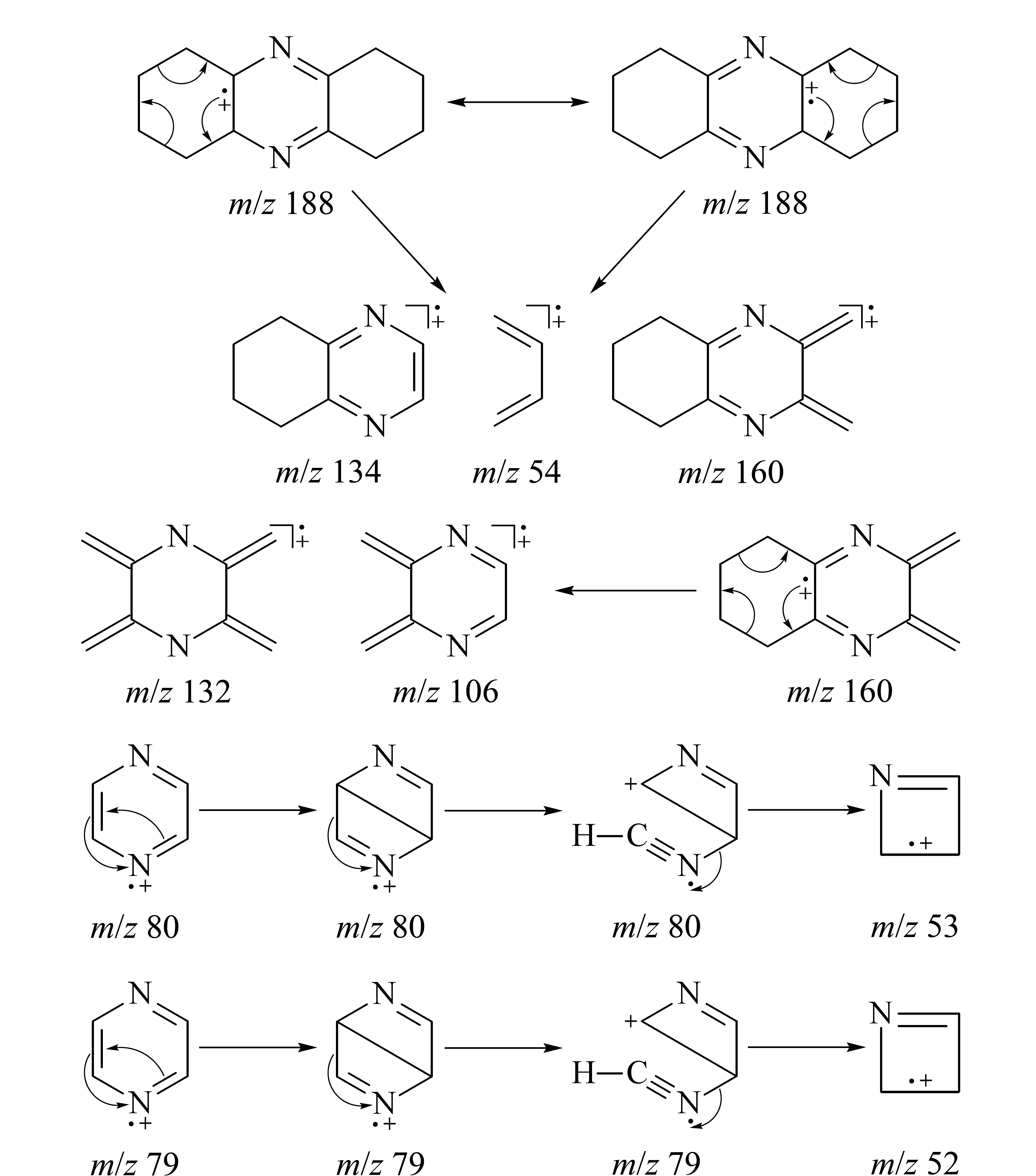
圖 5 組合工藝得到的CPL新雜質可能的電子轟擊裂解路線Fig. 5 Probable fragmentation pathways of the novel impurity in the CPL produced from the combined process
2.4 雜質來源分析
八氫吩嗪的來源與Beckmann重排反應緊密相關,目前已有多篇文獻[6-12]報道酸催化環己酮肟重排時將產生嚴重影響重排產物質量的副產物八氫吩嗪,如果后續分離裝置不能將八氫吩嗪有效脫除,產品的光密度值就可能超標,而且還會影響到產品的高錳酸鉀指標。
從八氫吩嗪本身的結構來看,其產生的方式有兩種,一種是α-氨基環己酮自身縮合并脫去一分子氫得到,α-氨基環己酮可由酸量不足時環己酮肟發生的Neber反應得到,這種物質本身極不穩定,極易在有堿存在時發生自縮合得到八氫吩嗪[15],這些反應需要的條件在組合工藝的操作條件中都具備。另一種是由鄰二環己酮和鄰二環己胺縮合得到。從組合工藝反應涉及的原料來看由后一種模式產生的可能性不大,因為在這個反應體系中雖會有鄰二環己酮,卻不存在鄰二環己胺。因此組合工藝中的八氫吩嗪來自于α-氨基環己酮自身縮合反應。而其反應前體則來自于環己酮肟的Neber重排反應,而不是一些文獻[6,10]中所述的環己酮肟的自縮合反應。事實上已有文獻[16]認為Beckmann重排產物和Neber重排產物是互為副產物的,對環己酮肟這種結構的原料更具備發生Neber重排的反應條件,而且Neber重排反應本身也是在深入研究Beckmann重排反應的過程中發現的。
在工業生產的粗產品出口P507工段取樣分析,檢測到了更高濃度的八氫吩嗪。對組合工藝本身而言,在P507工段樣品和最終產品中存在八氫吩嗪這種雜質有其合理性,因為P507正好是組合反應的終止階段,產物尚未進行任何精制,此時出現雜質的濃度最高點是正常的,最終產物中有該雜質是因為生產廠配置的精制手段對八氫吩嗪基本無效。但采用逆向法追尋雜質的產生點時,在組合反應發生前的配肟工藝中也發現了八氫吩嗪,配肟使用的溶劑環己烷回收自其他工段,其中含有硫酸和六氫苯甲酸這些甲苯法原料,在配肟過程中環己酮肟在六氫苯甲酸或者硫酸的作用下轉化成了八氫吩嗪。這與環己酮肟在酸量不足的情況下將發生副反應Neber重排的情況相吻合。
八氫吩嗪這種物質只有在CPL的雜質分析[11]和一些食物香料[17-20]的研究中才有提及,其中絕大部分都與Beckmann重排反應制備CPL有關。其首次合成報道于1954年[21],采用的原料就是α-氨基環己酮,熔點為112~113 ℃,沸點為135~170 ℃/1.5 kPa。1996年,Huang等[22]采用α-羥基環己酮與氨反應制備α-氨基環己酮的合成路線,同樣得到了八氫吩嗪,GC-MS分析其EI碎片為m/z188(豐度100,分子離子峰M+)、160(豐度39)、132(豐度15)、79(豐度12)和52(豐度16)碎片,這些信息對本工作中雜質的定性具有重要的參考價值。
3 結論
組合工藝得到的CPL中相對分子質量為188的雜質經GC-MS定性檢測為含氮化合物八氫吩嗪。在工業生產的P507工段取樣,GC-MS檢測到了更高濃度的八氫吩嗪。結合工藝過程及文獻報道分析了八氫吩嗪的產生過程,雜質結構的最終確定對己內酰胺制備工藝中選擇合適的精制路線具有重要的參考價值。
猜你喜歡
中國特種設備安全(2022年5期)2022-08-26 09:19:32
礦產綜合利用(2020年1期)2020-07-24 08:50:40
山東冶金(2019年6期)2020-01-06 07:45:54
收藏界(2019年2期)2019-10-12 08:26:06
世界農藥(2019年2期)2019-07-13 05:55:12
世界農藥(2019年2期)2019-07-13 05:55:10
模具制造(2019年3期)2019-06-06 02:11:00
山東工業技術(2016年15期)2016-12-01 05:30:59
銅業工程(2015年4期)2015-12-29 02:48:39
新疆鋼鐵(2015年3期)2015-11-08 01:59:52
