消腫片中楓香脂的鑒別與松香酸的檢查
2019-04-02 02:18:44黃海燕饒偉文鐘建理
中國藥業 2019年7期
黃海燕,饒偉文,鐘建理
(廣西桂林市食品藥品檢驗所,廣西 桂林 541001)
消腫片收載于《衛生部藥品標準·中藥成方制劑(第七冊)》,由楓香脂、沒藥、當歸、制草烏、地龍、乳香、馬錢子、香墨和五靈脂9味中藥組方,具有消腫拔毒功效,用于瘰疬痰核、流注、乳房腫塊、陰疽腫毒等[1]。方中楓香脂、乳香、沒藥均為樹脂類原料藥,且楓香脂為君藥,用量比較大。楓香脂質量參差不齊,市場摻雜、摻假現象嚴重,而乳香和沒藥為進口藥材,來源有限,價格較貴,市場上常有偽劣品出現[2-3]。偽劣楓香脂、乳香、沒藥藥材多為用松香摻假,含有松香酸,如誤用于中成藥生產中,將嚴重影響中成藥的質量。本研究中采用高效液相-二極管陣列(HPLC-DAD)法檢測5個廠家的5批消腫片樣品,結果檢出有松香酸的4批;同時,楓香脂薄層色譜(TLC)法鑒別試驗發現,結果僅有1批檢出楓香脂,其余4批未檢出。現報道如下。
1 儀器與試藥
1.1 儀器
Waters 2695型高效液相色譜儀(分離單元-Waters 2998 PAD檢測器,Waters公司);XS205DU型電子分析天平(梅特勒 -托利多公司);KQ-600E型超聲儀(昆山市超聲儀器有限公司);CAMAG AUTOMATIC TLC SAMPLER4全自動點樣儀,CAMAG TLC Visualizer薄層色譜照相儀,均購于CAMAG公司。
1.2 試藥
松香酸對照品(批號為111938-201201),楓香脂對照藥材(批號為121637-201201),均購于中國食品藥品檢定研究院;乙腈、甲酸為色譜純,其余試劑均為分析純,水為高純水;硅膠GF254預制薄層板(青島鼎康硅膠有限公司);消腫片(市售品,5批)。
2 方法與結果
2.1 楓香脂TLC法鑒別
取本品2片,研細,加甲醇20 mL,超聲處理20 min,濾過,濾液濃縮至2 mL,作為供試品溶液。按消腫片處方比例,制備不含楓香脂的陰性樣品,同法制成陰性對照品溶液。另取楓香脂對照藥材0.2 g,加甲醇10 mL,超聲處理20 min,取上清液作為對照藥材溶液。照TLC法試驗,吸取上述3種溶液各1 μL,分別點于同一硅膠GF254薄層板上,以石油醚 (60~90℃)-乙酸乙酯-冰醋酸(9 ∶1 ∶0.1)為展開劑,展開,取出,晾干,置紫外光燈(254 nm)下檢視。供試品溶液色譜中,在與對照藥材溶液色譜相應位置上顯相同顏色的斑點,表明陰性對照無干擾。詳見圖1。
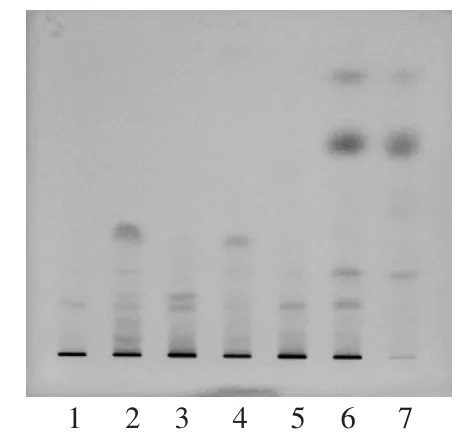
圖1 楓香脂薄層色譜圖
2.2 松香酸含量測定
2.2.1 色譜條件
色譜柱:AgilentZORBAX SB-C18柱(250 mm×4.6mm,5 μm);流動相:乙腈 -0.1% 甲酸溶液(82 ∶18);檢測波長:241 nm,DAD檢測器采集 200~400 nm光譜圖;柱溫:35 ℃;流速:1.0 mL /min。
2.2.2 溶液制備
取松香酸對照品約10 mg,精密稱定,置100 mL容量瓶中,加乙醇溶解并稀釋至刻度,精密量取1 mL置20 mL容量瓶中,加乙醇至刻度,制成每 1 mL含5 μg的對照品溶液。取本品研細,取約0.2 g,精密稱定,置具塞錐形瓶中,精密加入乙醇20 mL,稱定質量,超聲處理20 min,放冷,稱定質量,用乙醇補足減失的質量,搖勻,濾過,即得。
2.2.3 測定方法
精密吸取對照品溶液、供試品溶液各10 μL,注入液相色譜儀,以色譜峰的保留時間及DAD的紫外吸收圖譜進行定性鑒別。供試品溶液色譜中,在與對照品溶液色譜峰相應位置上不得出現相同的色譜峰,若出現保留時間相同的色譜峰,采用二極管陣列檢測器比較相應色譜峰的紫外-可見吸收光譜,吸收光譜應不同;若吸收光譜相同,則視為檢出松香酸。如檢出,則以外標法計算樣品中松香酸的含量。色譜圖見圖2和圖3。
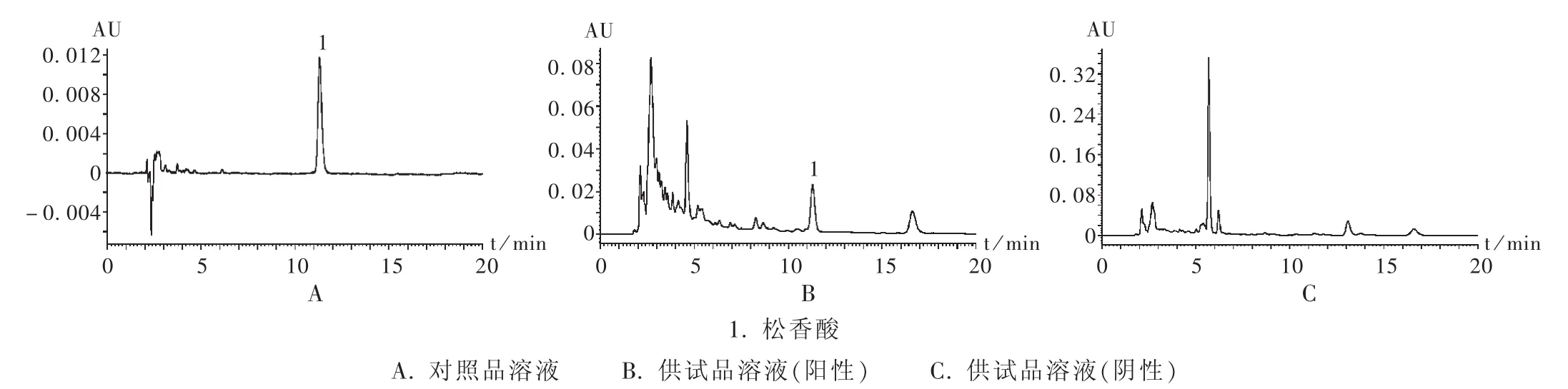
圖2 高效液相色譜圖
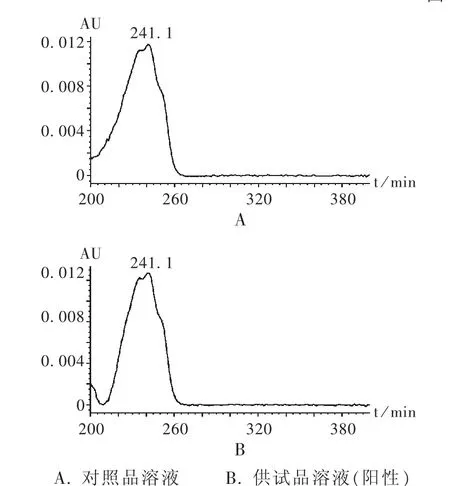
圖3 松香酸二極管陣列吸收圖
2.2.4 方法學考察
精密度試驗:取松香酸對照品溶液,連續進樣5次。結果松香酸峰面積的 RSD為0.86%(n=5),表明儀器精密度良好。
線性關系考察:取質量濃度為 5.053 6 μg/mL的松香酸對照品溶液,分別進樣 1,2,5,10,20,50 μL 測定。以峰面積(Y)為縱坐標、進樣量(X,μg)為橫坐標進行線性回歸,得回歸方程 Y=2.304 9 X-1.307 1,r=0.999 9(n =6)。結果表明,松香酸進樣量在 5.053 6 ~252.680 0 μg范圍內與峰面積線性關系良好。
空白試驗:按處方自制消腫片樣品,按2.2.3項下方法測定。結果未檢出松香酸,表明該方法無干擾。
穩定性試驗:取供試品溶液 1份,分別于0,2,4,8,12,24 h時測定,記錄峰面積。結果的 RSD為 0.34%(n=6),表明供試品溶液在24 h內穩定性良好。
重復性試驗:稱取同一批(1號)樣品6份,分別測定松香酸含量。結果平均含量為1.624 4 mg/g,RSD為0.77%(n=6),表明方法重復性良好。
加樣回收試驗:取已知松香酸含量的消腫片樣品(松香酸含量為 1.624 4 mg /g)約 0.1 g,共 9 份,分別加質量濃度為 0.051 8 g/L的對照品溶液 3 mL,為0.103 6 g/L 的 3 mL,為 0.051 8 g/L 的 10 mL,再分別精密加入乙醇20,20,10 mL,稱定質量,依法制備供試品溶液,測定松香酸的含量,計算回收率。結果見表1。

表1 松香酸加樣回收試驗結果(n=9)
檢出限和定量限測定:分別精密量取質量濃度為0.005 054 g/L 和 0.050 540 g/L 的松香酸對照品溶液各2 mL置錐形瓶中,再分別加入自制消腫片樣品0.203 3,0.202 6 g,依法制備供試品溶液,測定。結果松香酸色譜峰的信噪比(S/N)分別為4和13,方法的檢出限為 0.05 mg /g,定量限為 0.50 mg /g。
耐用性試驗:曾試驗不同色譜柱[DIKMA Inertsil ODS-3 柱(250 mm ×4.6 mm,5 μm),資生堂 CAPCELL PAK C18MG 柱(250 mm × 4.6 mm,5 μm)],以及液相色譜儀Agilent 1260型液相色譜儀(DAD檢測器),其測定結果均一致。
2.2.5 樣品含量測定
取不同廠家的樣品,依法制備供試品溶液,測定,按外標法計算含量。結果5批消腫片中有4批未鑒別出楓香脂卻檢出了松香酸,只有1批鑒別出楓香脂而且未檢出松香酸。詳見表2。

表2 5批樣品松香酸含量檢測結果
3 討論
3.1 展開劑選擇
曾比較了中國藥典中楓香脂鑒別項的展開劑正己烷-石油醚(60~90℃)-乙酸乙酯-冰醋酸(6∶2∶3∶0.2),但楓香脂的主斑點 Rf值偏高,故采用原國家食品藥品監督管理總局藥品檢驗補充檢驗方法和檢驗項目批準件(批準件編號為2016005)中相關楓香脂中松香酸檢查項補充檢驗方法的展開劑石油醚(60~90℃)-乙酸乙酯 -冰醋酸(9 ∶1∶0.1)[4];同時,楓香脂的主斑點 Rf值約為0.6,展開效果較理想,且陰性無干擾。
3.2 流動相選擇
曾參照楓香脂中松香酸檢查項補充檢驗方法試驗流動相乙腈-四氫呋喃-0.1%甲酸溶液(35∶25∶40),但楓香脂在松香酸色譜峰相近保留時間處出現干擾峰,不易分離。
3.3 檢測指標選擇
現已有很多對處方中含有乳香和沒藥等樹脂類原料藥的中成藥進行松香酸檢查[4-8]和補充方法如聶儉林等[9]進行了消腫片的質量標準研究,對當歸、地龍、五靈脂進行鑒別,對烏頭堿進行限量檢查,對士的寧進行含量測定,但尚無對消腫片進行松香酸檢查和楓香脂鑒別研究的文獻報道。本研究中收集5個廠家的5批消腫片,結果4個廠家的4批檢出了不該有的松香酸,應為楓香脂、乳香、沒藥原料藥帶入,同時這4批卻未檢出處方中為君藥的楓香脂,說明生產時未投料楓香脂或使用了假楓香脂。4批樣品中松香酸的含量測定結果相差也很大,說明其原料的摻假程度也各不相同。可見,消腫片生產廠家未能有效控制原料藥質量,市場上消腫片的質量參差不齊。本方法簡單、可靠,可用于提高消腫片的質量標準。
