桿狀體肌病新發突變臨床特征及基因診斷
2019-03-28 06:44:40牛煥紅成勝權
山西醫科大學學報 2019年3期
牛煥紅,成勝權
(空軍軍醫大學第一附屬醫院兒科,西安 710032;*通訊作者,E-mail:quanyi@fmmu.edu.cn)
桿狀體肌病(nemaline myopathy,NM)是一種罕見的先天性肌病,因在患者肌肉纖維中發現大量桿狀體結構而得名,發病率報道不一,呈常染色體顯性遺傳(AD)/常染色體隱性遺傳(AR)及散發。目前報道大多通過骨骼肌肌肉活檢病理診斷,肌細胞胞漿中的桿狀體是特征性肌肉病理改變[1]。本文報道的患兒經過基因檢測確診,該突變位點目前國內尚未見報道,且為新發突變,現將其臨床特征及其基因診斷特點總結如下,提高臨床醫生對本病的認識。
1 資料與方法
1.1 病例介紹
患兒,男,2歲3個月,主因發育遲緩、肌無力2年余于2018年5月就診我院。患兒于1月齡時即發現頸項軟,不能抬頭;1歲2個月時仍不會翻身、獨坐、爬行、站立,一直以腦性癱瘓診治;現回訪2歲6個月可扶站、扶走,智力語言發育大致正常;患兒系第2胎第2產,足月剖腹產,出生體重2.7 kg,否認窒息缺氧史。家族史無特殊記載。查體:體重7.5 kg,臉型瘦長,表情淡漠,脊柱后凸(見圖1),前囟閉合,頭圍47 cm,嘴唇稍厚,膨脹型嘴,高腭弓(見圖2),四肢肌力3級,肌張力低,雙側上、中、下腹壁反射(++),雙側肱二頭肌、肱三頭肌腱反射(+),雙側膝、跟腱反射未引出,雙側巴賓斯基征未引出。無震顫、手足徐動、舞蹈樣動作及感覺異常等。
輔助檢查:血、尿、便常規、肝腎功、心肌酶譜、離子五項、血糖、血氨、血乳酸、血尿有機酸代謝篩查、染色體核型、心臟彩超、頭顱核磁(MRI)均正常;雙側腓總神經、脛神經、正中神經、尺神經運動神經傳導速度(MCV)大致正常;雙眼視覺誘發電位(FVEP)、雙耳聽覺誘發電位(BAEP)未見異常;蓋塞爾智能發育測驗(1歲2個月時):動作能發育年齡(DA)2.8月、發育商(DQ)19.8分,應物能DA 8.7月、DQ 62分,言語能DA 9.3月、DQ 66分,應人能DA 10月、DQ 59分。
1.2 方法 基因測序
患兒生后不久即出現明顯的肌力、肌張力減低,充分告知患兒家長并簽署知情同意書后對患兒及其父母、姐姐各抽取3 ml全血,置EDTA抗凝管,送北京海思特醫學檢驗實驗室行基因測序,采用安捷倫外顯子芯片捕獲+高通量測序法,進行醫學外顯子5 000種疾病篩查。
2 結果
2.1 高通量測序結果
患兒1號染色體(chr1):229567822位點存在727(鳥嘌呤)G>(腺嘌呤)A的雜合突變,見表1。
2.2 sanger驗證結果
患兒一人存在c.727G>A突變,父親、母親、姐姐無突變(見圖3,4)。
2.3 對應數據庫報道疾病
該樣本在ACTA1基因外顯子區域發現一處雜合突變(見表2),該突變HGMD數據庫報道疾病為NM,家系驗證結果顯示該突變在父母雙方均未檢出,考慮為新發(De novo)突變。
表1桿狀體肌病患兒高通量測序結果

突變基因轉錄本Exon編號核苷酸變化氨基酸變化染色體位置深度測序ACTA1NM-001100exon5c.727G>Ap.Glu243Lyschr1-22956782251/86(0.63)
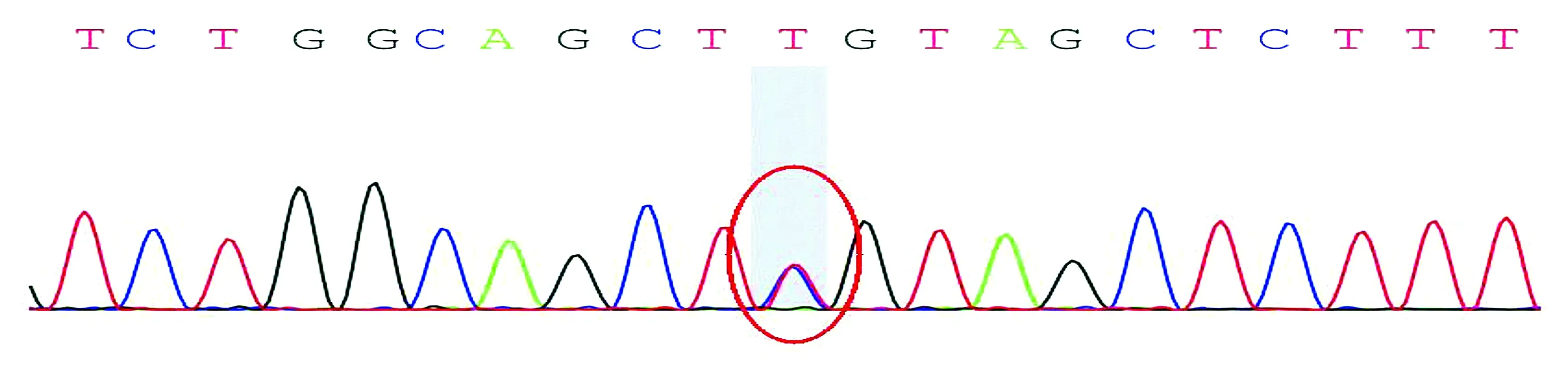
圖3 桿狀體肌病患兒sanger驗證
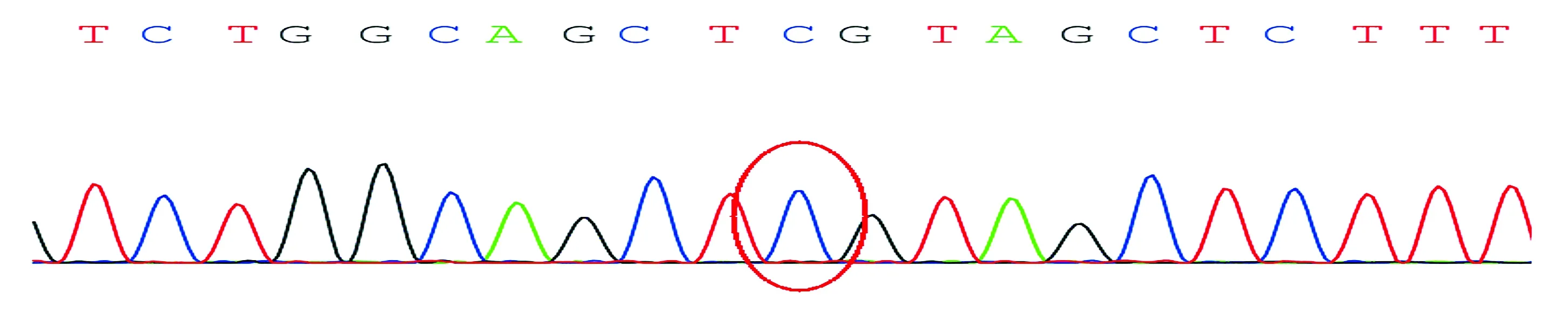
圖4 患兒父親sanger驗證
表2桿狀體肌病患兒HGMD專業版數據庫以及ACMD分級

基因突變位點HGMDpro報道生物學危害性ACMD分級ACTA1c.727G>ADM:桿狀體肌病可能有害(SIFT和Polyphen評估)-
3 討論
3.1 NM的致病基因
NM屬于罕見的先天性肌病,其主要病因為編碼細肌絲相關蛋白的基因突變[2]。目前已發現可引起NM的致病基因包括ACTA1、TPM3、TPM2、KBTBD13、NEB、TNNT1、CFL2、KLHL40、KLHL41、LMOD3、MYPN和MYO18B等10余種,NEB基因變異是NM最常見的原因,約占已發現突變基因的50%[3]。而ACTA1為NM第2常見致病基因,約占已發現突變基因的15%-25%[4],也是目前唯一已知與核內桿狀體相關的基因。ACTA1編碼骨骼肌α-肌動蛋白,該蛋白可存在于1型和2型肌纖維中,目前已發現200余種ACTA1突變,主要為錯義突變,多為AR,少數為AD。本文病例的ACTA1基因外顯子區域發現1處雜合突變,染色體定位1q42,致c.727G>A(鳥嘌呤>腺嘌呤),從而導致編碼α-肌動蛋白的氨基酸發生改變,p.Glu243>Lys(谷氨酸>賴氨酸),根據HGMDpro數據庫報道為致病,報告疾病為桿狀體肌病,家系驗證結果顯示該突變在父母雙方均未檢出,考慮為新發(De novo)突變,但不排除父親或母親存在生殖細胞突變嵌合體可能。根據SIFT和PolyPhen評估生物學危害性為可能有害,理論上此突變導致疾病發生的可能性大,可能為患者表型相關的致病突變。
3.2 NM的臨床特征
根據2000年歐洲神經肌肉病委員會國際協作組對NM的臨床分型,主要分為以下6型:①先天重癥型(16.1%):出生時即有嚴重的肌無力、肌張力低下,嚴重的呼吸功能障礙,多早期死亡,年齡不超過1歲。②先天中間型(20.3%):嬰兒期起病,出生時可有自主呼吸運動,但兒童早期可發展為無法自主呼吸運動,無法獨立行走和站立,11歲前借助輪椅是主要特征之一。介于先天重癥型和先天輕癥型之間。③輕癥型(經典型)(46.1%):嬰兒期/兒童期起病,主要以面肌、軀干肌無力,伴肌張力低下,運動發育遲緩,隨年齡增長肌力會有增加,病情相對穩定,多數患者的生活質量不受影響,智力、心肌收縮力多為正常。④兒童起病型(13.3%):與輕癥型類似,區別在于該型起病年齡為兒童晚期或青少年期。⑤成人起病型(4.2%):起病30-60歲,是一組異質性疾病,臨床表現和疾病進展都有很大的差異,多為散發,急性或亞急性起病,病程進展加重較快,易累及呼吸肌,多預后不良。⑥其他形式的NM:少見,表現為心肌病、眼肌麻痹、異常肌無力分布、核內桿狀體。根據患者致病基因及染色體定位的不同,所編碼的蛋白質及臨床表型不同,根據其對應的臨床表型有助于判斷其預后。如TPM2、CFL2基因突變,導致其分別編碼的β原肌球蛋白、人肌動蛋白素2發生改變,均可導致先天輕癥型NM發病;而ACTA-1、NEB、TPM3突變根據其染色體位點的不同所致NM可出現先天重癥型、輕癥型、其他各型等多種表型[2]。本文中患兒ACTA1基因突變,致其所編碼的α-肌動蛋白發生改變,導致臨床表型為輕癥型,隨年齡增長肌力增加,現可扶站扶走,智力與同齡兒相仿,預后較好。
3.3 NM的診斷與鑒別診斷
3.3.1 診斷 該病的診斷主要依靠骨骼肌肌肉活檢,無論哪種致病基因所致,其特征性表現均為肌細胞胞質中可見高密度的桿狀體,MGT染色可以見到最為典型的紅染桿狀體,不同部位的肌肉組織及不同肌纖維中桿狀體數目不盡相同,故取材不同可能會漏診,需二次或多次取材。尤其對于綜合醫院的患兒,由于條件限制肌肉活檢很難實施,因肌肉活檢術為有創檢查且術后患兒活動受限、傷口護理及恢復需要時間。隨著分子生物學技術的發展與應用,肌肉活檢不再是唯一診斷的金標準,將會有越來越多的NM通過基因檢測被報道。
3.3.2 鑒別診斷 臨床對于有肌無力的患兒需要鑒別的疾病較多,如進行性肌營養不良、皮肌炎、脊髓性肌萎縮、腦性癱瘓等等,應根據其臨床病史及查體做出初步定位定性判斷,對于像該患兒出現全身軟性癱瘓者,定位為下運動神經元及其以下病變,如能排除神經傳導與神經肌肉接頭處病變如重癥肌無力后,考慮定位診斷位于脊髓前角的運動神經元或肌肉時,二者臨床很難鑒別,若肌電圖能發現巨大電位、纖顫電位、失神經電位、高幅多相運動電位,可能為運動神經元病;若發現短暫、低幅多相運動電位可能為肌肉病變,然而,要做到這一點實屬不易,需要高年資有豐富經驗的兒童肌電圖醫生來實現,最終二者的鑒別需要骨骼肌病理活檢或相應的基因檢測結果方可確診。
本病尚無有效治療方法,有研究[5]報道,采用馬法蘭或者免疫抑制劑如環磷酰胺和甲基強的松治療能改善部分病人的臨床癥狀,但尚未經過大規模試驗。隨著分子生物學技術的發展與應用,NM的發病機制研究以及基因治療將可能成為新的研究熱點。
