二苯甲烷雙馬來酰亞胺二元芳香胺固化劑的合成及其固化動力學研究
2019-03-25 15:16:39張思思高念李文翔夏蘭君管蓉
粘接 2019年5期
張思思 高念 李文翔 夏蘭君 管蓉


摘要:以二苯甲烷雙馬來酰亞胺(BDM)、4,4-二氨基二苯甲烷(DDM)為原料,采用Michael加成法制得環氧樹脂(E-44)固化劑(BDM-DDM),通過紅外光譜(FT-IR)和液質聯用(LC-MS)手段驗證了所得產物為目標產物,用非等溫DSC法探究了BDM-DDM與E-44固化體系的固化過程,結果表明:非等溫差示掃描量熱(DSC)法能夠確定BDM-DDM/E-44固化體系的固化工藝。利用Kissinger方程和Crane方程進行計算可得到固化反應的反應活化能E.為49.92KJ/mol和反應級數n為0.98。
關鍵詞:BDM;DDM;固化劑;固化動力學
中圖分類號:TQ314.256文獻標識碼:A 文章編號:1001-5922(2019)04-0008-04
使用環氧樹脂需配合固化劑,固化劑對環氧樹脂體系的宏觀性能起著重要作用。現有的環氧樹脂固化劑不能滿足實際應用的需要,因此,有必要對其進行改性來拓展環氧樹脂的使用范圍。目前,環氧樹脂固化劑使用相對最廣泛的為多元胺類固化劑,其中具有代表性的是4,4-二氨基二苯甲烷(DDM)和4,4-二氨基二苯醚(DDE),其缺點是難以在常溫下與環氧樹脂形成均相體系,自身熔點較高,固化困難,在很大程度上限制了其應用范圍。為此,改性多元胺類固化劑是當前研究的熱點之一,常見的方法有環氧樹脂加成改性、邁克爾加成反應、曼尼希加成反應、硫脲或酮類加成反應。
雙馬來酰亞胺(BMI)分子中含有酰亞胺環結構,因此,在耐熱性和力學性能方面表現較優,而且BMI具有較好的可塑性和流動性,可采用與環氧樹脂一樣的加工方式,且具有優異的耐熱性、耐腐蝕性和吸濕率較小等特點。二苯甲烷雙馬來酰亞胺(BDM)是一種常用的雙馬來酰亞胺,由于分子中苯環的存在,使得BDM具有較高的熔點,而本身的對稱結構也使BDM具有較好的結晶性,保障了其良好的熱穩定性和優異的力學性能。在傳統的二元胺分子,如DDM、DDE中引入BMI剛性基團是一種較好的改性途徑,此舉不僅可以提高環氧樹脂固化物的力學性能和耐熱性,而且引入BMl分子結構之后混合體系的交聯度降低,固化物的韌性提高。
本研究以環氧樹脂(E-44)、二元芳香胺固化劑(DDM、BDM)為原料,通過Michael加成合成了雙馬來酰亞胺固化劑BDM-DDM,采用傅里葉變換紅外光譜(FT-TR)和液質聯用(LC-MS)對產物結構進行了表征。利用非等溫差示掃描量熱(DSC)法研究了BDM-DDM/E-44固化體系的固化動力學,確定了合適的固化工藝。
1實驗部分
1.1實驗原料
環氧樹脂(E-44),工業級,岳陽石化股份有限公司;二苯甲烷雙馬來酰亞胺(BDM),分析級,上海笛柏化學品技術有限公司;4,4-二氨基二苯甲烷(DDM),分析級,山東佰仟化工有限公司;1,2-二氯乙烷、三乙胺、甲醇、無水乙醇,分析級,國藥集團化學試劑有限公司。
1.2儀器與設備
DSC200F3型差示掃描量熱(DSC)儀,德國耐馳有限公司;Spectrum one傅里葉變換紅外光譜儀(FTIR),美國Pekin-Elmer公司;TGA2型熱重差熱分析儀(TG),瑞士梅特勒-托利多公司;液質聯用儀(LC-MS),安捷倫科技有限公司。
1.3雙馬來酰亞胺固化劑的合成及應用
將一定量的DDM溶于1,2-二氯乙烷并加入到三口燒瓶中,65℃水浴鍋中攪拌溶解,然后通過恒壓滴液漏斗加入DDM物質的量1/2的1,2-二氯乙烷BDM溶液,同時分3次加入催化劑三乙胺,滴加完之后保溫3h。反應結束之后減壓蒸餾,抽去反應體系中的溶劑,將收集到的產物用無水乙醇多次沖洗并用索氏提取法提取24h,70℃下真空干燥12h即可得到產物BDM-DDM。
將一定量的E-44加入燒杯中,70℃水浴攪拌,再加入一定量的BDM-DDM,攪拌均勻后,接著加入DDM,在70℃下真空脫泡5~10min,得到混合良好的E-44固化體系,倒入涂有脫膜劑并預熱好的模具中,按照設定好的固化工藝參數進行固化,自然冷卻即可得到E-44固化物。
1.4性能測試
(1)微觀結構特征:采用傅里葉變換紅外光譜(FT-IR)儀進行表征BDM和BDM-DDM(分別取少量原料及合成的固化劑試樣均勻地涂在溴化鉀晶片上,掃描次數為32次,掃描波數范圍為400~4000cm-1)。
(2)物質分析:采用液質聯用(LC-MS)儀測試BDM-DDM的圖譜(取少量的樣品溶于甲醇試劑中)。
(3)熱性能:采用熱重差熱分析(TG-DSC)儀測量E-44固化劑體系固化過程的放熱量(升溫速率分別為2.5、5、7.5、10℃/min,升溫范圍為30~300℃,N2環境,流速為40mL/min)。
2結果與討論
2.1FT-IR表征
BDM與BDM-DDM的FT-IR譜圖如圖1所示。由圖1可知:3521~3270cm-處為-NH2的伸縮振動特征吸收峰,3107cm-1、3105cm-1處為苯環上C-H的伸縮振動特征峰,1614cm-1處為新出現的-NH的彎曲振動特征吸收峰,3000~2800cm-1處的峰為-CH2-的伸縮振動特征吸收峰,這是酰亞胺環加成后的結果;BDM-DDM譜圖中690cm-1處并沒有峰出現,這是因為酰亞胺環上的C=C雙鍵發生了加成反應,同時,BDM-DDM譜圖中在1706cm-1左右的峰得到了保留,可以說明反應并未影響雙馬來酰亞胺基。
2.2LC-MS驗證
圖2為BDM-DDM的LC-MS圖。由圖2可知:199.1258為DDM的分子質量,表明合成產物中有少量原料分子殘留,其中755.2270為BDM-DDM的分子質量。結合FF-IR圖譜和LC-MS圖譜分析表明,合成產物為目標產物BDM-DDM。
2.3固化體系固化工藝的確定
為了得到合適的E-44固化工藝,采用DSC分別測量BDM-DDM/E-44體系在升溫速率為2.5℃/min、5℃/min、7.5℃/min和10℃/min時的DSC曲線,實驗結果如圖3所示。由圖3可知:不同速率下固化反應的起始固化溫度(Ti)、峰值溫度(Tp)和終止固化溫度(Tf)各不相同,其數據列于表1中,其中,△H為固化反應的放熱量。
特征溫度T與升溫速率β的關系如圖4所示。由圖4可知:外推至β=0時的溫度參數Ti、Tp、Tf分別為74℃、122℃和217℃。由于BDM-DDM分子間作用力較大,低溫又會使體系黏度增大,反應速度變緩,因此初始固化溫度應較高一點。又由于BDM-DDM固化劑的分子質量比較大,使得單位體積的活性基團較少,反應活性偏低,所以適當延長高溫固化的時間對體系固化完全有利。本研究選取的BDM-DDM/E-44體系固化工藝確定為,120℃/2h+140℃/2h+200℃/4h。
2.4固化動力學
E.表示反應的活化能,代表反應進行的難易程度,也反映了對溫度的敏感程度。Ea越大,說明反應越難進行,以及對溫度越敏感。本研究采用非等溫DSC分析法確定了BDM-DDM/E-44體系的固化反應動力學參數。
2.4.1Kissinger法
Ea可由Kissinger方程(式1)求得:
式中:β為升溫速率(K/min);L表示峰頂溫度(K);Ea表示活化能(J);R代表理想氣體常數,數值為8.314J/(mol·K)。Kissinger法相對簡單,且與反應級數n無關。
BDM-DDM/E-44固化體系在不同蝦的動力學參數列于表2中。將不同升溫速率下的-ln(β/T2p)對100/Tp)作圖,并擬合直線(如圖5所示),由直線的斜率可求得活化能Eao由擬合曲線圖5可知:擬合的線性相關性較好,說明此法較為準確。
2.4.2Ozawa法
Ozawa法不需知道具體的固化機理函數即可求出Ea,推導過程如下。
式中,F(X)表示與α有關的函數。
當α一定時,將lgβ對1/Tp作圖,并進行線性擬合,將直線斜率代入式(3):
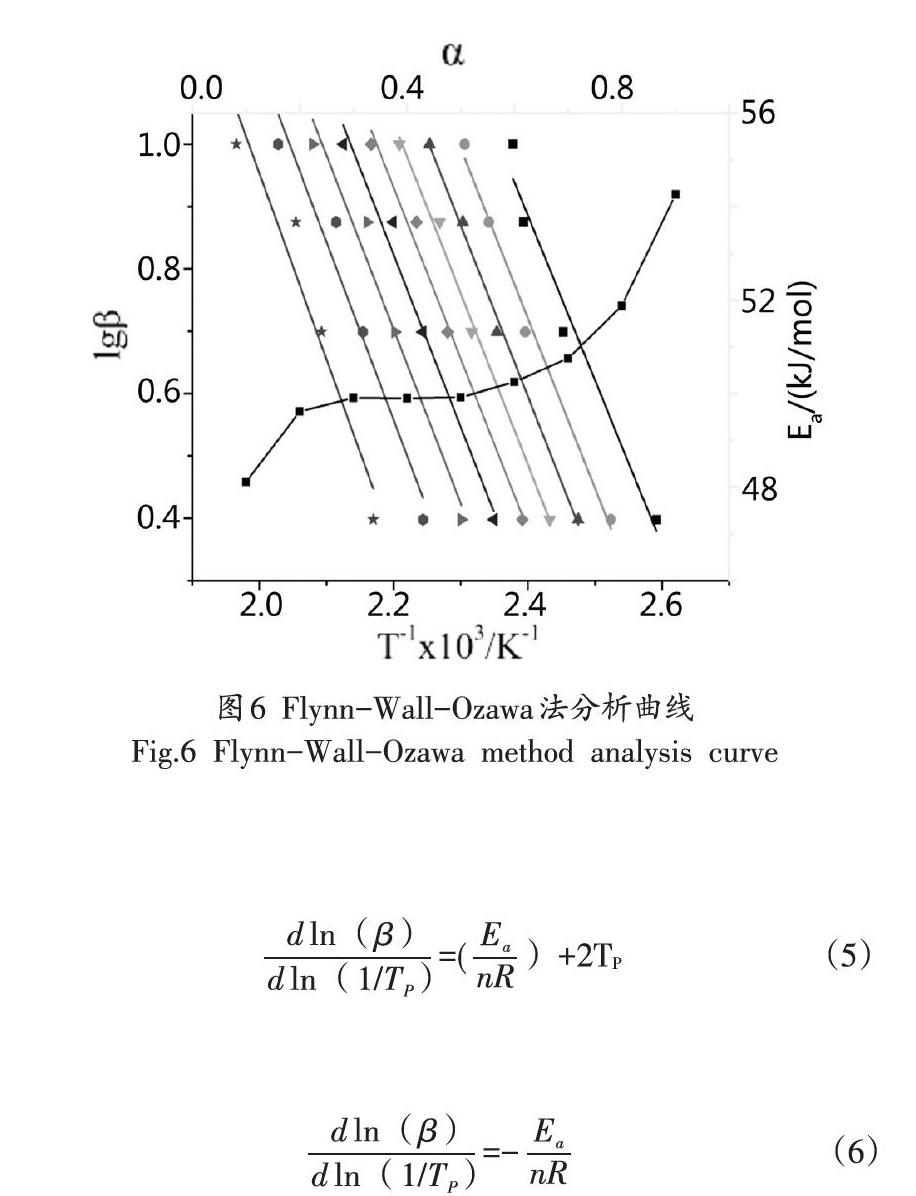
即可求出活化能Eao固化體系的Ozawa法擬合曲線如圖5所示,由圖5可知:擬合曲線的線性相關性良好。
由Kissinger方程和Ozawa方程計算出的活化能Ea和線性相關系數列于表3中。其中Ek是由Kissinger法計算出的活化能,E0是由Ozawa法計算出的活化能。
FWO模型避開了機理函數帶來的誤差,計算結果較為準確。由FWO方程可知:Ea、α、T和β之間有式(4)的關系式。
當β一定時,將lg(β)對1/T作圖,并線性擬合,Ea可由擬合直線的斜率求出。FWO法的分析曲線如圖6所示。由圖6可知:擬合直線的線性相關性良好。本研究取α為0.5,體系活化能Ea為49.92kJ/mol。與芳雜環結構的環氧/芳香二胺類樹脂體系相比,DDM-BDM/E-44體系的活化能處于較低的水平,這表示,引入BDM結構后體系的固化反應活性并沒有發生改變。
本研究采用Crane方程來求解反應級數,根據Crane方程可得式(5)表達式。
又因為對熱固性樹脂而言,有Ea>2Tp,故式(5)可簡化為式(6)。
式中,n為反應級數。
將FWO法與Kissinger法計算得到的BDM-DDM/E-44體系的E.取平均值,以ln(β)對1/Tp作圖,并擬合直線,如圖6所示,由直線可求得斜率,即可求出α為0.98。
3結論
本研究成功合成了改性固化劑BDM-DDM,采用非等溫DSC法確定BDM-DDM/E-44體系的固化工藝為120℃/2h+140℃/2h+200℃/4h,并利用Kissinger和Crane等方程計算得到了固化反應的反應活化能Ea=49.92kJ/mol和反應級數n=0.98。
