鮮繭與干繭生絲的結構與性能比較及其鑒別方法
2019-03-25 06:58:42蓋國平董永春陳興燦
紡織學報 2019年3期
李 冰, 蓋國平, 郭 蔚, 董永春, 陳興燦
(1. 廣西出入境檢驗檢疫局 檢驗檢疫技術中心, 廣西 南寧 530021; 2. 天津工業大學 紡織科學與工程學院, 天津 300387)
目前繅絲企業所繅制的生絲主要包括鮮繭生絲和干繭生絲。鮮繭生絲通常是指將收購鮮繭冷凍干燥并在特定溫度的水中真空滲透或煮繭處理后直接繅絲而制成的生絲;而干繭生絲則是采用傳統高溫烘干技術處理并在較高溫度中煮繭而得的生絲。由于鮮繭生絲在生產過程不需要高溫烘繭和煮繭等工藝,能耗低,且鮮繭比干繭更易抽絲,因此其生產效率高,工人勞動成本低,已近成為生絲加工的發展趨勢[1]。此外,鮮繭繅絲企業還可以充分利用鮮繭制絲時產生的副產品蠶蛹,以獲得較高的經濟利潤。與干繭生絲相比,鮮繭生絲具有生產成本低,生產效率高等優勢。在繅絲成本日益高漲和冷凍儲存成本降低的背景下,越來越多的繅絲企業選擇繅制鮮繭生絲[2],但由于鮮繭生絲較低的價格,使得以鮮繭生絲冒充干繭生絲進行交易而謀求高利潤的不良現象時有發生。此外,目前繅絲企業通常不會在生絲產品包裝上直接標注生絲的鮮或干繭性質,且2種生絲外觀形貌幾乎相同,這使得絲綢企業在采購原料時難以辨識而導致使用不當的織造工藝,直接影響蠶絲織物生產效率和產品質量;因此,關于生絲表面性能的研究以及鑒別技術的建立十分重要。
目前已經出現了關于生絲性能比較和鑒別方法的報道。黃繼偉等[3]發現,與干繭生絲相比,鮮繭生絲含膠率和白度較高,但抱合力較差,二者均以無規卷曲和β折疊的結晶結構為主。章琪超等[4]則認為鮮繭生絲較粗糙,初期絲膠溶失率高,而結晶度低。喬鐵軍等[5]研究表明,在鮮/干繭生絲的抱合實驗中,鮮繭生絲的抱合力比干繭生絲差。而實踐證明基于生絲強度、白度或含膠率等方面差異的鑒別方法準確性和普適性較差。此外,目前也出現了基于紅外光譜或高效液相色譜技術的鑒別方法[6],但是都存在鑒別過程復雜,成本高且時間長,難以滿足企業需求的問題。
為此,本文借助掃描電子顯微鏡、傅里葉紅外光譜儀、X射線衍射儀、熱重分析儀和表面接觸角測試等手段對2種生絲的結構與性能進行了表征和比較,然后基于2種生絲在特定表面活性劑水溶液中沉降時間的顯著不同建立了一種更加準確和簡單的鑒別技術,以期為生絲商檢、生絲質量指標完善以及絲綢企業選擇正確的織造工藝等提供更加科學的理論依據,以促進我國繅絲和絲綢制造技術向低成本和高性能方向發展。
1 實驗部分
1.1 材料與試劑
27種生絲樣品來自廣西繅絲企業,其規格均為22.22~24.44 dtex;溴化鉀、丙酮、十二烷基苯磺酸鈉、十二烷基磺酸鈉、聚氧乙烯辛基苯酚醚-10,均為分析純試劑,購于天津市科密歐化學試劑有限公司。
1.2 實驗儀器
Nicolet Magna-560型傅里葉紅外光譜儀(美國尼高力儀器公司);PHI 5600型X射線光電子能譜儀(美國珀金埃爾默公司);S-4800型場發射掃描電子顯微鏡(日本日立公司); STA 409 PC型熱分析儀(德國耐馳公司);DSA100 型光學接觸角測量儀(德國克呂士公司);HWCL-1型恒溫磁力攪拌器(鄭州長城儀器有限公司)。
1.3 生絲的表面形貌觀察
使用丙酮對2種生絲樣品進行洗滌處理,2 min后取出烘干,然后使用導電膠將伸直生絲黏附在樣品臺上,并在其表面噴金處理后放入干燥器中靜置12 h,最后在20.0 kV電壓條件下使用掃描電子顯微鏡(SEM)觀察和比較其表面形貌差異。
1.4 生絲的化學組成和微結構分析
將生絲樣品與一定量的溴化鉀混合后壓片制樣,使用傅里葉紅外光譜儀(FT-IR)在分辨率為4 cm-1和掃描20次的條件下對樣品的化學組成進行分析。
使用X射線衍射儀(XRD)對樣品表面的晶體結構進行分析。測試條件:輻射源為CuKα,波長為0.154 nm,管電壓和電流分別40 kV和100 mA,掃描范圍為3°~40°,掃描速度為2(°)/min。
1.5 生絲的熱性能表征
使用熱分析儀測量生絲樣品從室溫升溫到800 ℃過程中的質量損失率,得到其熱失重(TGA)和微商熱失重(DTG)曲線。測試條件:樣品質量為(1.0±0.01)mg,環境介質為氮氣,流速為20 mL/min,升溫速率為5 ℃/min。
1.6 生絲表面接觸角測試
在溫度為(20±1)℃和相對濕度為(65±5)%的條件下使用接觸角測量儀測定生絲樣品的表面接觸角。首先將經丙酮處理的樣品以伸直狀態固定在樣品臺上,然后通過脈沖將超純水液滴噴射到樣品表面上,在10 s內拍下液滴與樣品接觸的全過程,并采用量角法計算單纖維的靜態接觸角。
1.7 生絲的鑒別實驗
生絲預處理工藝:在室溫條件下首先使用丙酮洗滌處理生絲樣品2 min,取出后再使用蒸餾水對其水洗3次后晾干。然后將其置于溫度為20 ℃和相對濕度為65%的環境中平衡12 h以上備用。
測試液的配制:稱取一定質量的十二烷基苯磺酸鈉、0.05 g有機硅消泡劑和200 mL蒸餾水添加到250 mL燒杯中,將它們混合均勻并靜置2 h后待用。
生絲樣品的制備:將一定根數經過上述預處理的生絲纖維整齊碼好,并用1根相同生絲進行捆扎打結,隨后量好絲束長度并用剪刀裁剪成2.5 cm的長度,即制得生絲樣品。
測試方法:在室溫環境條件下,使用干凈的鑷子將上述捆扎的生絲樣品平行地輕放于測試液的液面中央(避免與燒杯壁接觸影響沉降)并同時開始計時,記錄生絲樣品最終沉降觸到燒杯底部的時間。
2 結果討論
2.1 生絲的表面形貌分析
圖1示出干繭生絲和鮮繭生絲的掃描電鏡照片。可以看出,2種生絲均由多根繭絲平行排列而成,且表面附著的絲膠將繭絲黏著在一起形成束狀結構。比較2種生絲表面形貌發現,干繭生絲較鮮繭生絲表面附著的絲膠膜更厚、更均勻,連續性也更好。在干繭生絲結構中,繭絲之間的縫隙幾乎都被絲膠填滿,繭絲之間排列得更加緊密。而在鮮繭生絲結構中,繭絲之間的縫隙明顯變大,其中存在的絲膠較少。此外,鮮繭生絲表面附著細小絲膠顆粒的現象更加明顯,這主要與2種生絲繅絲工藝方面的差異有關。具體而言,鮮繭生絲在繅絲過程中未經過高溫煮繭,繭絲表面附著的雜質顆粒不能完全從其表面脫落溶解到水中,且包覆在絲素外面的絲膠也沒有得到充分膨潤,使得鮮繭生絲繅絲上膠時絲膠的黏合力度不及干繭生絲,因此纖維表面顆粒多和繭絲之間空隙大。這表明2種生絲纖維在形態結構上有差異,可在鑒別時參考,但是由于掃描電鏡測試成本較高,因此在實際應用中受到一定的限制。

圖1 干繭生絲和鮮繭生絲的掃描電鏡照片Fig.1 SEM images of two kinds of raw silk. (a)Dry cocoon raw silk(×1 000);(b)Fresh cocoon raw silk(×1 000);(c)Dry cocoon raw silk(×1 800);(d)Fresh cocoon raw silk(×1 800)
2.2 生絲的化學結構分析

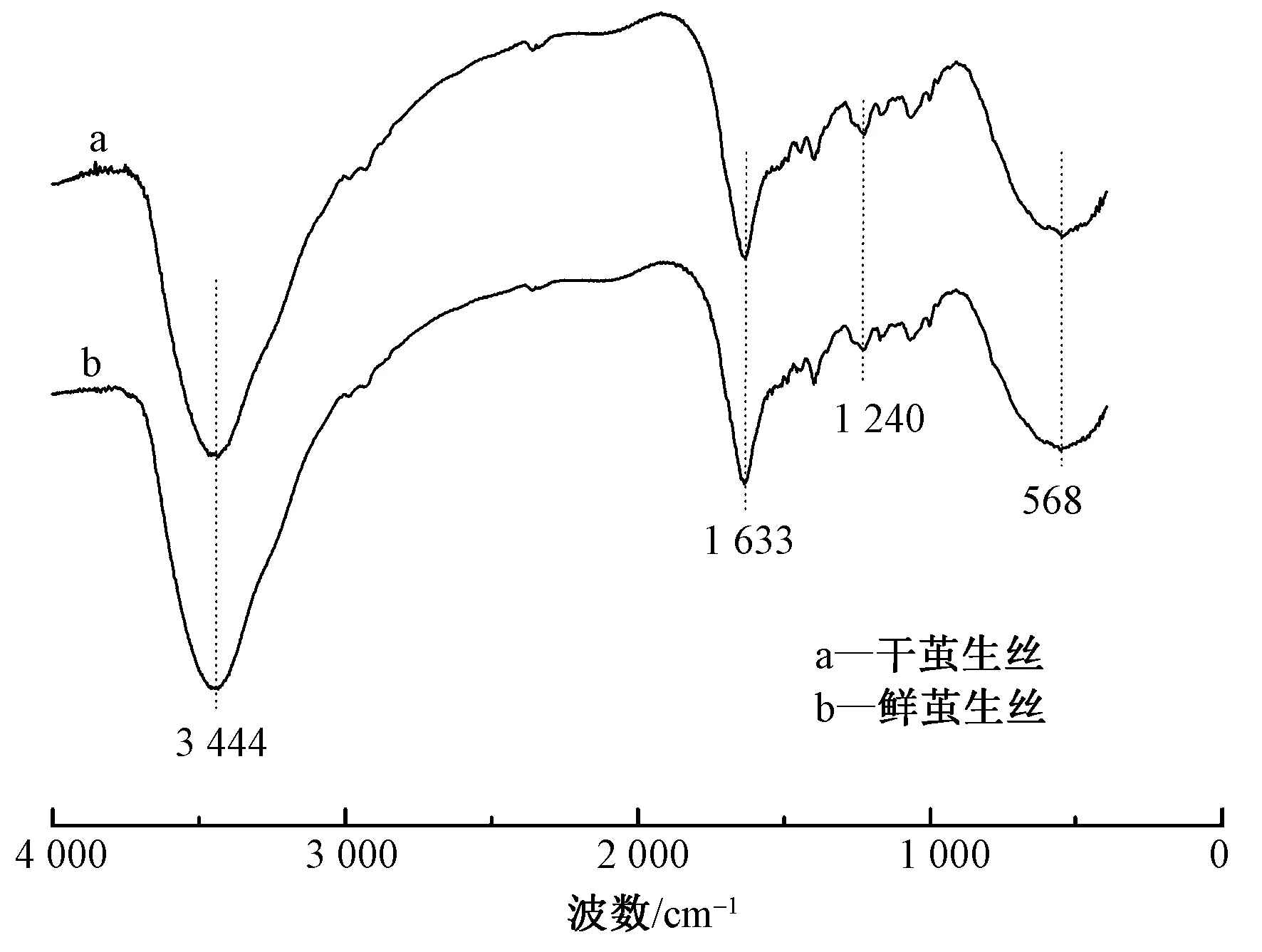
圖2 鮮繭生絲和干繭生絲的紅外光譜圖Fig.2 FT-IR spectra of fresh cocoon silk and dry cocoon silk
2.3 生絲的晶體結構分析
使用X射線衍射儀對鮮繭生絲和干繭生絲表面的晶體結構進行測定,得到2種纖維的XRD圖譜,并參照Hermans等[10]提出的方法利用Peakfit軟件對其進行分峰擬合處理, 以考察二者在結晶度和晶粒尺寸等方面的差異,分峰擬合處理結果如圖3所示。
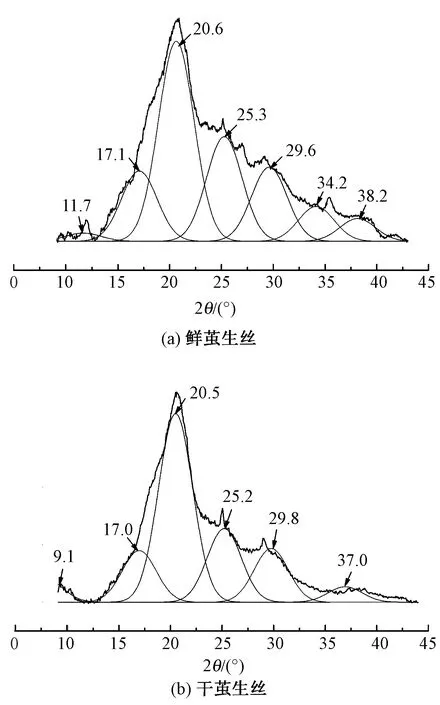
圖3 鮮繭生絲和干繭生絲的XRD譜圖Fig.3 XRD spectra of fresh cocoon silk (a) and dry cocoon silk (b)
絲素蛋白的結晶形態可分為Silk I型和Silk II型2種晶體結構。Silk I型是一種趨向于α螺旋的結構,而SilkⅡ型則是一種趨向于反向平行β折疊鏈的結構[11-13],絲素蛋白分子的構象和結晶形態在不同條件下也會有所改變。在蠶絲纖維中,Silk I型和SilkⅡ型2種晶體結構通常是同時存在的,且盡管Silk II型 較 Silk I 型結構相對穩定,但是在一定的化學反應條件下這2種構象也可以相互轉化。據文獻[3,14]報道,Silk I型結構的主要衍射特征峰分別在2θ為12.2°(中強)、19.7°(強)、24.7°(中強)、28.2°(中強)、32.3°(弱)、36.8°(中弱)、40.1°(中弱)處;而SilkⅡ型結構的主要衍射特征峰在2θ分別為9.1°(中強),18.9°(中強)和 20.7°(很強)、24.3°(弱)、39.7°(中弱)處。圖3中的XRD譜線經過分峰擬合處理后可以清楚地看到鮮繭絲在2θ為11.1°、17.1°、20.6°、25.3°、29.6°、34.2°和38.2°處存在衍射吸收峰,干繭絲則在9.1°、17.0°、20.5°、25.2°、29.8°和37.0°處存在衍射吸收峰,二者在20.6°左右的衍射吸收峰均有很高的峰值,該特征峰與SilkⅡ型結構的特征衍射吸收峰相對應。這表明鮮繭絲與干繭絲蛋白中同時存在α螺旋結構和β折疊結構,且以SilkⅡ型(β折疊結構)的晶體結構為主[15]。根據圖中曲線計算得到鮮繭絲結晶度為38.85%,干繭絲結晶度為47.04%。這表明鮮繭生絲的結晶度略低于干繭生絲,因此可通過2種生絲纖維的結晶度來輔助判定其鮮干繭絲性質。但2種生絲纖維的結晶度相差值不穩定,所以不能僅通過結晶度來區分。
2.4 生絲的熱性能分析
生絲熱分解反應的質量損失過程主要分為3個階段,如圖4所示。鮮繭生絲和干繭生絲的第1個質量損失階段是溫度從室溫升到125 ℃的階段,在該階段鮮繭生絲和干繭生絲的質量減少分別為10%和8%左右,且微商熱質量損失在63 ℃左右達到峰值,此時2種生絲的熱分解曲線趨勢基本一致,但鮮繭生絲的質量減少速率略高于干繭生絲,這是因為此階段主要對應于生絲纖維中自由水、結合水以及纖維表面小分子有機溶劑等的揮發,鮮繭生絲在繅絲之前未經過高溫烘干,因此纖維中存在較多的自由水和結合水。在第2質量損失階段2種生絲纖維分解速率最快,其從200 ℃開始逐漸發生質量損失,并在305.5 ℃達到峰值,該階段終止溫度約為450 ℃,因此鮮繭生絲和干繭生絲在200~450 ℃質量損失過程中其質量減少分別為 59%和57.5%,此時鮮繭生絲的質量損失速率略高于干繭生絲,其主要原因是該質量減少階段對應著生絲纖維中氨基酸鏈的分解和結晶區的分裂[16],最終導致纖維發生熱分解。由上述XRD譜圖可知,鮮繭生絲的結晶度低于干繭生絲,因此導致其質量減少速率略高于干繭生絲。第3質量損失階段2種生絲纖維的TGA和DTG曲線基本一致,該階段主要是纖維分子裂解和炭化階段,此時鮮繭生絲和干繭生絲的最終質量減少約為70% 和69%。由此可以看出,鮮繭生絲和干繭生絲的熱分解曲線盡管稍有不同,但其趨勢基本一致,因此并不能通過熱質量損失曲線來區分這2種生絲纖維。
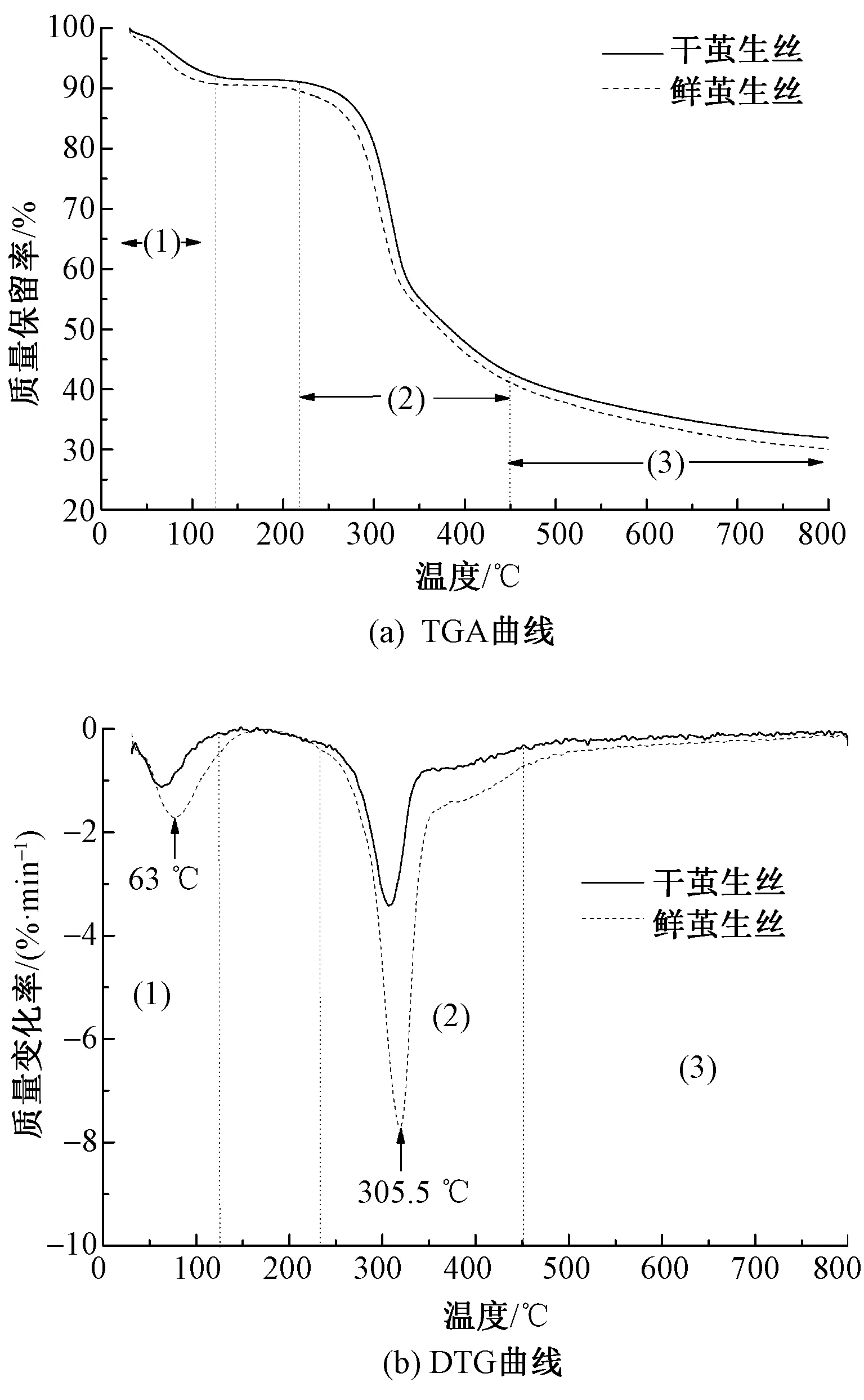
圖4 鮮繭生絲和干繭生絲的熱分析曲線Fig.4 Thermal analysis curves of fresh cocoon silk and dry cocoon silk. (a)TGA curves;(b)DTG curves
2.5 生絲的表面接觸角比較
使用接觸角測量儀測試了多組鮮繭生絲和干繭生絲對水介質的接觸角,結果如圖5和表1所示。
由圖5和表1看出,干繭生絲的水接觸角為48°~53°,而鮮繭生絲的水接觸角為31°~39°,二者相差約15°,表明鮮繭生絲比干繭生絲具有更好的親水性能。這主要與鮮繭生絲中繭絲之間的縫隙較大以及表面相對粗糙有關。此外,干繭生絲在繅絲前經過高溫烘繭,這會導致油脂從繭蛹外層溢出并被吸附到繭絲表面,使得干繭生絲表面的親水性變差。

圖5 鮮繭生絲和干繭生絲的水接觸角比較Fig.5 Comparison on water contact angle of fresh cocoon raw silk b (a) and dry cocoon raw silk g (b)
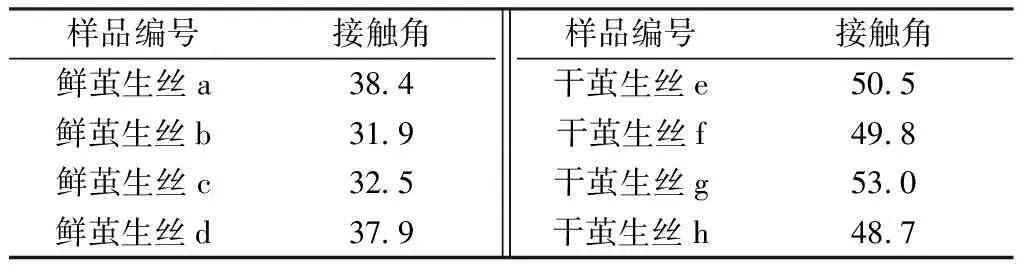
Tab.1 Comparison on water contact angle of two kinds of raw silk(°)
2.6 生絲鑒別方法的建立
根據2種生絲表面親疏性能方面的差異,建立了測定生絲束在特定表面活性劑水溶液的沉降時間的方法,以有效地鑒別2種生絲。為更加顯著地增加2種生絲在沉降時間上的差別以提高鑒別的準確性,本文考察了表面活性劑的性質、濃度以及生絲束樣品中生絲根數對其沉降時間的影響。
2.6.1表面活性劑種類對生絲沉降時間的影響
分別使用2種陰離子型表面活性劑十二烷基苯磺酸鈉(SDBS)與十二烷基磺酸鈉(SDS)和1種非離子型表面活性劑聚氧乙烯辛基苯酚醚-10(OP-10),配制濃度均為0.5 mmol/L的水溶液,然后將20根生絲組成的2種生絲束樣品分別放置于溶液表面并測定它們的沉降時間。結果表明:當SDBS存在時,鮮繭生絲的沉降時間為17 s,干繭生絲的沉降時間則大于600 s;當SDS存在時,鮮繭生絲和干繭生絲的沉降時間均大于1 800 s;當OP-10存在時,鮮繭生絲的沉降時間為34 s,干繭生絲的沉降時間則大于150 s。
當使用SDBS時,2種樣品的沉降時間差別巨大,其中鮮繭生絲樣品的沉降時間遠小于干繭生絲,說明鮮繭生絲樣品更易被SDBS水溶液潤濕和浸透,表1也已證明鮮繭生絲比干繭生絲具有更好的親水性能。而當使用SDS時,2種樣品的沉降時間都很長,無法用來鑒別2種生絲束樣品。當使用OP-10時,盡管2種樣品的沉降時間也有顯著差別;但是遠不如使用SDBS時2種樣品的沉降時間差別大,因此,有必要進一步研究SDBS濃度對沉降時間的影響,以獲得最佳實驗條件。
2.6.2SDBS濃度對生絲沉降時間的影響
為考察SDBS濃度對2種生絲沉降時間的影響,首先配制不同濃度的SDBS水溶液,然后將不同數量生絲構成的生絲束樣品分別放置于溶液表面并測定它們的沉降時間,結果見表2。
從表2看出:當水中未添加SDBS時,2種樣品在600 s內均未出現沉降現象;當溶液中SDBS濃度為0.50、1.0 mmol/L時,鮮繭生絲樣品的沉降時間均在50 s以內,而干繭生絲樣品的沉降時間均已超過600 s;當其濃度增加至2.5 mmol/L時,鮮繭生絲樣品的沉降時間仍低于50 s,干繭生絲樣品中由4~20根生絲組成的其沉降時間仍超過600 s,而超過20根生絲組成的2個樣品的沉降時間縮短顯著,尤其是由36根生絲組成樣品的沉降時間僅為53 s,已接近由相同根數鮮繭生絲組成樣品的沉降時間(49 s);當其濃度為3.50~5.0 mmol/L時,鮮繭生絲樣品的沉降時間并未發生顯著變化,而干繭生絲樣品的沉降時間隨著樣品中生絲根數的增加而逐漸下降,但是未超過相應鮮繭生絲樣品的沉降時間。這說明水溶液中SDBS的添加對2種生絲樣品的沉降時間具有顯著影響。對于鮮繭生絲樣品,較低濃度(0.50、1.0 mmol/L)的SDBS就會導致其沉降時間明顯下降,而SDBS濃度的提高并未引起其沉降時間顯著縮短。對于鮮繭生絲樣品,低濃度(0.50、1.0 mmol/L)的SDBS不會導致其沉降時間發生顯著降低,而SDBS濃度的提高則引起其高根數樣品沉降時間顯著變短,且這種趨勢隨著SDBS濃度的提高變得更加突出。導致這種現象的主要原因是鮮繭生絲比干繭生絲具有更好的親水性能,其更易于被SDBS水溶液浸透而沉降;而干繭生絲表面親水性較差,需要較高濃度的SDBS水溶液才能使其浸透而沉降。此外,鮮繭生絲樣品的根數對其沉降時間的影響并沒有明顯的規律性,當其根數為20時樣品的沉降時間通常最短。與其不同的是,隨著干繭生絲樣品根數的增加,其沉降時間逐漸變短,尤其在SDBS濃度提高時這種變化趨勢更顯著,這種差異仍然與2種生絲表面親水性不同有關。

表2 2種生絲束樣品在不同濃度SDBS溶液中的沉降時間Tab.2 Settling time of two kinds of raw silk in SDBS solutions with different concentrations
根據上述2種生絲樣品表面親水性的差異以及表面活性劑性質和濃度的影響作用結果,能夠建立一種基于二者在特定濃度SDBS水溶液中沉降時間的有效鑒別方法,其中建議SDBS濃度為0.50~1.0 mmol/L,樣品根數不多于20根,這是因為在此條件下2種樣品沉降時間的差異最大,提高了鑒別方法的準確性。通常在此條件下,沉降時間大于600 s的樣品被認定為干繭生絲,否則為鮮繭生絲。
2.7 生絲鑒別方法的驗證
從生絲生產企業收集26種商品化生絲樣品(鮮繭生絲和干繭生絲各13種),首先對其進行前處理,然后采用上述方法測定它們的沉降時間,并根據實驗結果進行鮮繭、干繭的判定,結果如表3所示。
表3數據顯示:所有鮮繭生絲樣品的沉降時間相對較短,大多數不超過100 s,其與實驗結果相符的準確率達到100%;而10種干繭生絲樣品的沉降時間高于600 s,3種樣品的沉降時間在180~230 s之間,其準確率接近77%。值得說明的是,對于3種測試不準確的樣品,其沉降時間明顯長于鮮繭生絲樣品,也可基本判斷為干繭生絲樣品;但是需要使用其他技術進一步考察。基于以上分析看出,采用此方法鑒別鮮繭生絲樣品的準確性明顯高于干繭生絲樣品。
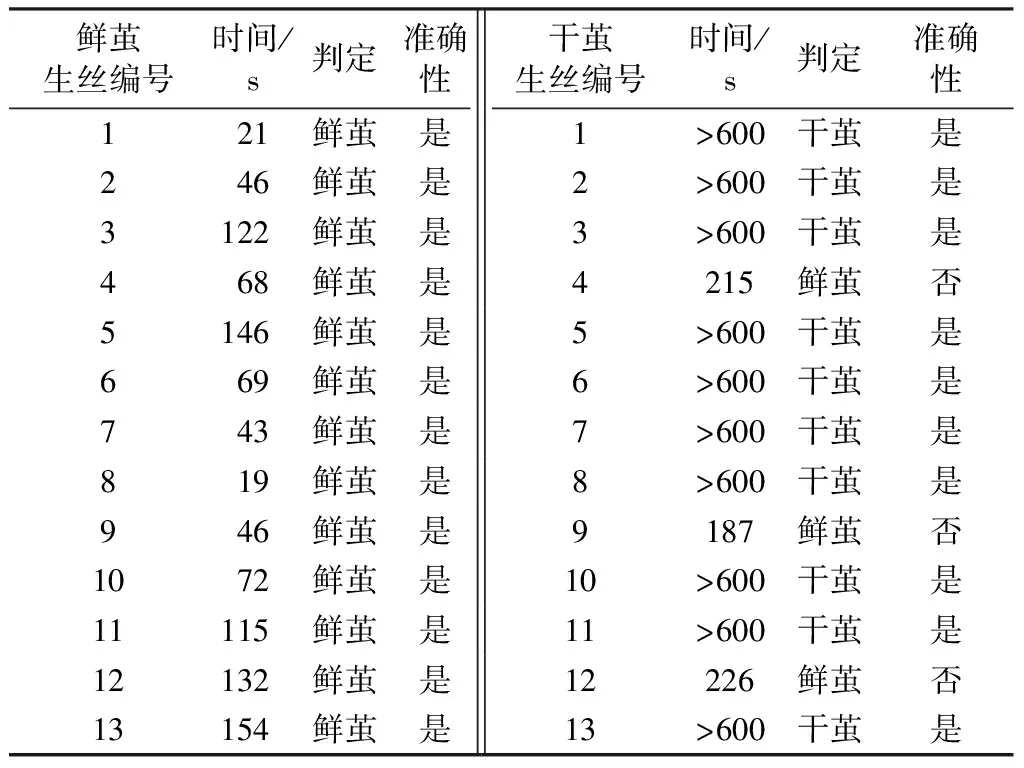
表3 26種商品化生絲樣品的沉降時間Tab.3 Settling time of 26 kinds of commercial raw silk
3 結 論
1)鮮繭生絲的表面形貌與干繭生絲相比,前者的表面顆粒較多且繭絲之間空隙也較大。鮮繭生絲和干繭生絲大分子二級構象主要為β折疊構象,鮮繭生絲的結晶度略低于干繭生絲。鮮繭生絲和干繭生絲的熱分解曲線盡管稍有不同但其趨勢基本一致。
2)鮮繭生絲比干繭生絲具有更好的親水性能,二者水接觸角相差約15°。根據2種生絲表面親疏性能方面的差異,建立了測定生絲束在特定表面活性劑水溶液的沉降時間的方法以有效地鑒別2種生絲。最終確定該方法的最佳工藝參數:測試液中十二烷基苯磺酸鈉的濃度為0.5~1.0 mmol/L,捆扎的纖維根數在20以內。采用該方法對26種鮮繭生絲和干繭生絲樣品進行鑒別驗證,其鑒別鮮繭生絲的準確率能達到100%,鑒別干繭生絲的準確率達到77%。
FZXB
