靈芝多糖對ApoE-/-動脈粥樣硬化小鼠TLR4/NF-κB信號通路的影響?
2019-03-18 01:58:40楊燕燕謝金東周建華俞春英劉德強王訓立
中國中醫基礎醫學雜志 2019年1期
楊燕燕,謝金東,周建華,俞春英,林 瑋,劉德強,王訓立
(福建中醫藥大學,福州 350122)
動脈粥樣硬化(atherosclerosis,AS)是一種復雜的多因素疾病,表現為動脈壁對各種炎癥損傷的炎癥-增殖反應[1-2]。目前,AS防治措施有多種選擇,經皮腔內冠狀動脈成形術雖能緩解AS心肌缺血癥狀,但容易發生術后再狹窄;他汀類西藥可迅速、有效的治療AS,但作用單一,長期應用會導致明顯的不良反應;而多種中藥也被用于防治AS疾病,并提示具有一定程度的療效[3]。近年來,靈芝的抗AS作用機制研究多數是基于大鼠動物模型進行的血脂監控及病理形態學分析[4],但靈芝多糖(ganoderma lucidum polysaccharides, GLPs)治療AS涉及炎癥因子和免疫反應的分子機制尚未見報道。因此,本研究基于ApoE-/-小鼠AS模型和TLR/NF-κB信號通路,分析GLPs對TLR4及其下游MyD88依賴性或非依賴性信號通路的影響,探討其對AS干預作用的分子生物學基礎。
1 材料
1.1 實驗動物與分組
8~10周齡ApoE-/-小鼠50只,按隨機數字表法分為模型組、GLPs低劑量組(200 mg/kg·d)、GLPs中劑量組(500 mg / kg·d)、GLPs高劑量組(800 mg / kg·d)及辛伐他汀組(1.8 mg / kg·d)各10只,8~10周齡C57BL/6小鼠10只,設為正常對照組。小鼠購自北京維通利華實驗動物技術有限公司(許可證號SCXK(滬)2012-0002),喂飼于福建中醫藥大學實驗動物中心(許可證號SYXK(閩)2014-0005)SPF級動物實驗室。
1.2 主要儀器與試劑
美國ABI 2720 PCR擴增儀;美國賽默飛世爾17R小型高速冷凍離心機;美國ABI Stepone plus型熒光定量PCR儀;美國Biorad Mini-PROTEAN@Tetra小型垂直電泳槽;美國Biorad Power Pac電泳儀;美國Biorad ChemiDoc XRS +凝膠成像分析系統;普通二級清潔飼料,高脂飼料(1%膽固醇,10%豬油,89%基礎飼料);生物工程(上海)股份有限公司SK1312 Total RNA Extractor試劑盒;生物工程(上海)股份有限公司SK2445第一鏈cDNA合成試劑盒;生物工程(上海股份有限公司)B639273 2X SG Fast qPCR Master Mix(High Rox);GLPs購自杭州眾芝康菇生物科技有限公司;TLR4、NF-κB、MyD88等抗體購自美國CST公司;TRAF6、TRIF等抗體購自英國Abcam公司;TRAM抗體購自德國Merck Millipore公司;Pierce Goat Anti-Mouse IgG(H+L)-HRP Antibody和Pierce Goat Anti-Rabbit IgG-HRP Antibody購自美國Thermo Fisher公司。
2 方法
2.1 AS動物模型制作
正常組小鼠喂飼普通飼料,ApoE-/-小鼠給予高脂飼料,連續喂養共90 d。造模后期按照各藥物干預組設定劑量每日灌胃給藥1次,正常組和模型組給予等量生理鹽水,治療期為30 d。解剖取病變主動脈血管,驗證AS疾病模型造模成功與否并進行后續實驗。
2.2 實時熒光定量PCR
表1顯示,血管組織總RNA提取步驟嚴格按照說明書進行,并通過反轉錄反應體系得到cDNA,參照Gene Bank,由生工生物工程(上海)股份有限公司合成。PCR反應總體系20.0 μL:2×SybrGreen qPCR Master Mix 10.0 μL,Template cDNA 2.0 μL,10 μM PCR Forward Primer 0.4 μL,10 μM PCR Reverse Primer 0.4 μL,ddH2O 7.2 μL。反應條件為:95 ℃ 3 min,95 ℃ 7 s,55 ℃ 10 s,72 ℃ 15 s,共45個循環。以GAPDH為內參,數據計算依據2-ΔΔCT法[5]。

表1 各基因的引物序列及擴增產物的長度
2.3 Western Blot
提取動脈總蛋白,基于BCA法定量配制上樣蛋白樣品,SDS-PAGE垂直板全濕法電泳;轉于PVDF膜上,封閉液室溫封閉1 h,加入1∶1 000 稀釋的一抗,4 ℃孵育過夜,TBST洗滌;加入1∶3 000稀釋的HRP標記二抗室溫孵育1 h,洗滌后ECL顯色;經凝膠成像系統檢測,分析各組目標條帶的灰度值,計算相對表達量。
2.4 統計學方法
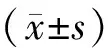
3 結果
3.1 ApoE-/-小鼠AS模型建立
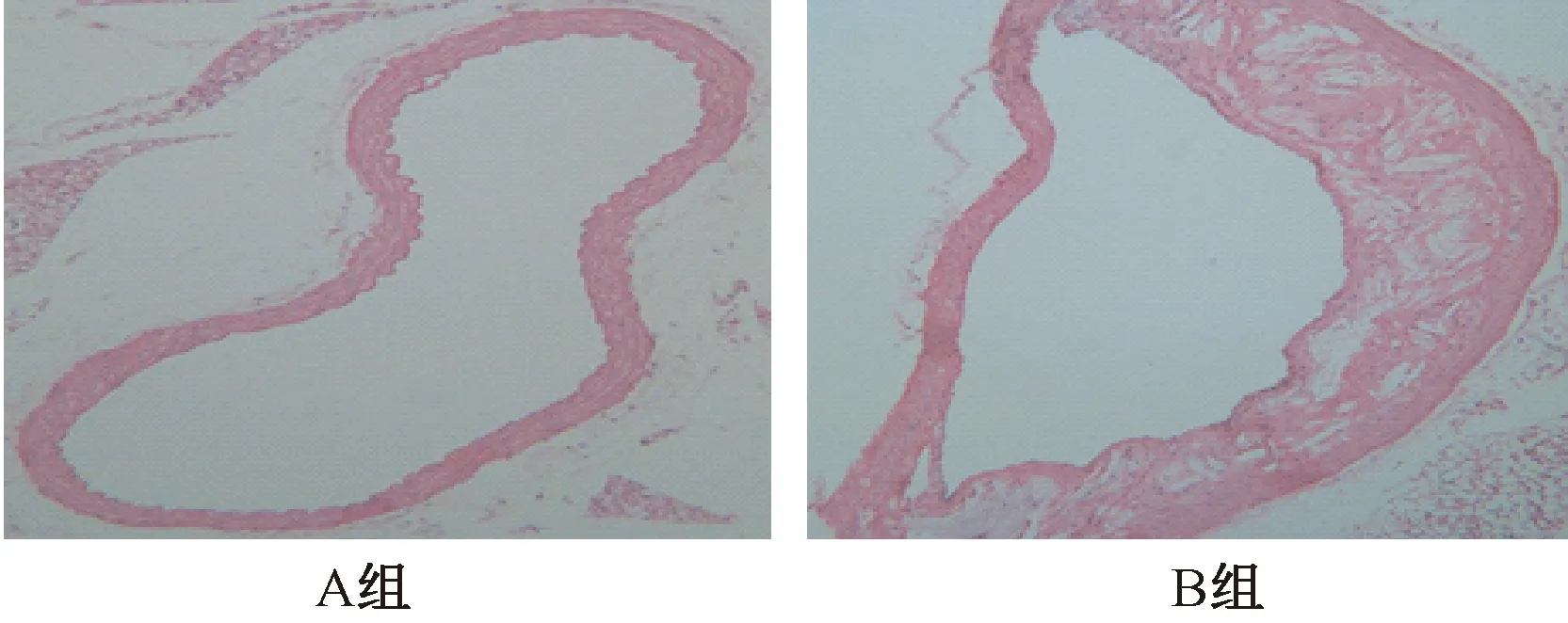
圖1 小鼠動脈病理組織學比較
圖1表2顯示,正常組主動脈壁層次清晰,內膜連續,中膜平滑肌細胞排列整齊,未見AS斑塊形成。模型組則顯示主動脈管壁厚薄不均,血管內膜增厚,內部脂質池增大,管腔內斑塊沉積,有泡沫細胞、炎癥細胞浸潤,表現為典型的AS病理特征。同時血清血脂水平檢測顯示,模型組TC、TG、LDL-C水平顯著升高,而HDL-C顯著降低,并具有統計學意義。基于上述證實,ApoE-/-小鼠AS模型建立成功。
3.2 RNA提取及降解狀況
圖2顯示,小鼠動脈組織總RNA條帶的18S、28S條帶清晰完整,基本無降解。

表2 正常組和模型組小鼠血脂水平比較
注:與正常組比較:*P<0.05,**P<0.01
3.3 靈芝多糖對TLR4及其下游MyD88信號通路元件表達的影響
3.3.1 TLR4、NF-κB、MyD88和TRAF-6 mRNA表達 表3顯示,與正常組比較,模型組TLR4(P<0.05)、NF-κB(P<0.05)、MyD88(P<0.05)和TRAF-6(P<0.01)mRNA表達顯著升高。與模型組比較,經藥物干預后各治療組TLR4、NF-κB、MyD88和TRAF-6 mRNA表達均有不同程序的降低。其中西藥對照組效果最為明顯,TLR4、MyD88及NF-κB mRNA比較差異有統計學意義(P<0.05),TRAF-6 mRNA差異有統計學意義(P<0.01)。而中藥GLPs治療組與模型組比較則表現為低劑量組NF-κB、中劑量組MyD88和高劑量組TLR4、NF-κB、MyD88 mRNA比較差異有統計學意義(P<0.05);同時GLPs各劑量組的TRAF-6 mRNA差異有統計學意義(P<0.01)。

圖2 RNA檢測電泳結果
3.3.2 TLR4、NF-κB、MyD88和TRAF-6蛋白表達 表3顯示,各組的蛋白相對表達趨勢與mRNA基本一致。與正常組比較,模型組TLR4(P<0.01)、NF-κB(P<0.05)、MyD88(P<0.05)和TRAF-6(P<0.01)蛋白表達顯著升高。與模型組比較,經藥物干預后各治療組各指標蛋白表達均有不同程序的降低,其中西藥對照組最為明顯,TLR4、MyD88及NF-κB差異有統計學意義(P<0.05),TRAF-6差異有統計學意義(P<0.01)。而中藥GLPs治療組與模型組比較,則表現為高劑量組TLR4和中、高劑量組的NF-κB、MyD88蛋白表達差異有統計學意義(P<0.05),同時GLPs各劑量組的TRAF-6比較差異有統計學意義(P<0.01)。
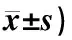
表3 各組小鼠TLR4及其下游MyD88依賴性通路元件mRNA和蛋白表達水平比較(相對表達量,
注:與正常組比較:*P<0.05,**P<0.01;與模型組比較:#P<0.05,##P<0.01
3.4 靈芝多糖對MyD88非依賴性信號通路元件表達的影響
3.4.1 TRAM和TRIF mRNA表達 表4顯示,與正常組比較,模型組TRAM(P<0.05)和TRIF(P<0.01)mRNA表達水平顯著升高,差異有統計學意義。與模型組比較,經藥物干預后各治療組TRAM和TRIF mRNA表達均有下降趨勢,西藥對照組效果較為明顯(P<0.05),但GLPs各劑量組比較差異無統計學意義(P> 0.05)。
3.4.2 TRAM和TRIF 蛋白表達 表4顯示,各組TRAM和TRIF蛋白相對表達趨勢與mRNA基本一致。與正常組比較,模型組TRAM(P<0.05)和TRIF(P<0.01)蛋白表達水平顯著升高。與模型組比較,經藥物干預后各治療組TRAM和TRIF蛋白均具有下降趨勢,西藥對照組效果較為明顯(P<0.05),但GLPs各劑量組比較差異無統計學意義(P> 0.05)。
4 討論
TLR4貫穿于AS起始、進展、斑塊不穩定及斑塊破裂等各個時期[6]。MyD88依賴性途徑表現為介導IRAK-1和IRAK4結合TLR4,IRAK-1自磷酸化后解離、募集并活化下游TRAF-6,隨后活化TAK1,激活IKK復合體,誘導IκB磷酸化并釋放NF-κB轉移至核內,經胞內級聯反應啟動相關基因轉錄,介導機體內早期的天然免疫及炎癥反應[7-9]。本實驗結果顯示,與正常組比較,模型組小鼠TLR4及下游MyD88依賴性信號通路主要細胞因子TLR4、NF-κB、MyD88及TRAF-6 mRNA和蛋白相對表達量均顯著升高。采用GLPs干預后,各指標表達均有不同程度下降趨勢,差異有統計學意義。因此,推測GLPs對TLR4及下游MyD88依賴性信號通路有一定程度的拮抗作用,對TLR4介導的TLR4/MyD88/NF-κB通路可能具有阻斷作用。而各劑量組的TRAF6表達水平與模型組比較下降趨勢最為顯著,這可能是由于TRAF6處于TLR信號通路介導的細胞凋亡和炎癥活化反應的中心點,起著至關重要的橋梁作用。這與AS發病機制的主流學說“炎癥學說”相一致[10]。
表4 各組小鼠MyD88非依賴性通路元件mRNA和蛋白表達水平比較(相對表達量,
注:與正常組比較:*P<0.05,**P<0.01;與模型組比較:#P<0.05,##P<0.01

注:1.正常組;2.模型組;3.辛伐他汀對照組;4.靈芝多糖低濃度組;5.0靈芝多糖中濃度組;6.靈芝多糖高濃度組圖3 各組小鼠TLR4、NF-κB、MyD88和TRAF-6蛋白表達比較

注:1.正常組;2.模型組;3.辛伐他汀對照組;4.靈芝多糖低濃度組;5.靈芝多糖中濃度組;6.靈芝多糖高濃度組圖4 各組小鼠TRAM和TRIF蛋白表達比較
TLR4還可不依賴MyD88激活IRF-3、IRF-7和NF-κB,通過相關接頭分子TRAM作用于TRIF,隨后結合TRAF6并活化IKK復合體,致使IκB泛素化,最終NF-κB釋放至細胞核,一系列炎癥反應由此產生[11]。而當前研究提示,MyD88非依賴途徑僅存在于TLR3和TLR4通路中[12-13]。本實驗結果顯示,與正常組比較,模型組小鼠TRAM、TRIF mRNA及蛋白表達明顯升高,采用GLPs干預后指標表達有所降低,但差異無統計學意義。因此,推測GLPs對TLR4介導的MyD88非依賴性信號通路可能無阻斷作用。
在AS療效方面,中藥治療組的療效不如西藥對照組,并未顯示出明顯的濃度依賴性。這與中藥在適當劑量時起效慢、療程長的現象相吻合。GLPs空間構型復雜,本身即具有雙向調節作用,而此調節作用主要受藥物劑量、配伍、炮制加工、藥物代謝動力學等因素的影響。因此,GLPs適宜劑量還需通過調整相應用藥量、用藥時間及方式來加以改善。與此同時,GLPs發揮免疫功能和抗炎效用的分子機制目前尚無具體定論[14-16]。其抗AS分子機制除TLRs/TLR4之外,可能還存在同時與多種受體結合,激活多條信號通路相互作用,而多糖與受體親和力的差異又將導致不同傳導通路對細胞激活效應的不同。因此,各種細胞表面模式識別受體的相互拮抗或協同調控機制仍需進一步的研究。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
課堂內外·初中版(科學少年)(2023年10期)2023-12-10 00:43:06
全科護理(2022年10期)2022-12-26 21:19:15
國際放射醫學核醫學雜志(2021年10期)2021-02-28 08:41:58
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
學苑創造·A版(2020年9期)2020-10-13 09:41:02
數學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
