止痛化癥膠囊的HPLC 指紋圖譜研究
2019-03-08 01:42:12吳福林董慶海譚靜王涵林紅強劉金平李平亞
特產研究 2019年1期
吳福林,董慶海,譚靜,王涵,林紅強,劉金平,李平亞
(吉林大學藥學院,長春 130021)
止痛化徵膠囊由黨參、炙黃芪、炒白術、丹參、當歸、雞血藤、三棱、莪術、芡實、山藥、延胡索、魚腥草、北敗醬、蜈蚣、全蝎、土鱉蟲、泡姜、肉桂等共19 味中藥組成,主治益氣活血、散結止痛。用于氣虛血瘀所致的月經不調、痛經、癓瘕,癥見行經后錯、經量少、有血塊、行經小腹疼痛、腹有癓塊,慢性盆腔炎見上述證候者。近年來,有關止痛化癥膠囊的研究主要集中在臨床應用及有關化學成分的含量測定[1-4],未見對其有關制劑質量控制標準、藥效物質基礎及入血成分等的研究報道。中藥指紋圖譜[5-8]是控制天然藥物質量的有效方式之一,能全面整體地反映藥品的質量,為中藥的質量評價和控制提供參考。本研究首次利用高效液相色譜(HPLC)法對止痛化癥膠囊進行指紋圖譜研究,建立了方法穩定、簡便、可靠的止痛化癥膠囊高效液相色譜指紋圖譜。結合指紋圖譜對不同批次止痛化癥膠囊相似度進行評價,為止痛化癥膠囊的鑒別、質量控制提供新的科學依據,保證藥品有效性及安全性,為制定全面的止痛化癥膠囊質量控制方法奠定一定工作基礎。
1 材料
1.1 儀器
1525型HPLC、Waters 2998 二極管陣列檢測器(American Waters 公司);FA1104N 型萬分之一電子分析天平(上海菁華科技儀器有限公司);R201D 恒溫水浴鍋、旋轉蒸發器(上海豫康科教儀器設備有限公司);KQ3200V 型超聲波清洗儀(昆山市超聲儀器有限公司)。
1.2 試劑
乙腈、甲醇(色譜級,美國Fisher 試劑公司);水(純凈水,杭州娃哈哈集團有限公司);其他試劑均為分析純。
1.3 藥品及對照品
止痛化癥膠囊由吉林金寶藥業股份有限公司提供,10 批藥品批號依次為160303、161002、160905、161001、160502、160501、160503、160904、161003、160701。
丹參素(151106)、原兒茶酸(171109)、黨參炔苷(17062207)、紫草酸(171220)、丹酚酸B(150927)(北京普天同創生物科技有限公司);迷迭香酸(11871-2001102)、原兒茶醛(110810-200205)(中國食品藥品檢定研究院)。
2 方法與結果
2.1 色譜條件
色譜柱:UniSil?5-120 C18 Ultra(4.6 mm×250 mm,5m);流動相:乙腈-水(含0.05%的甲酸);流速:1.0 mL/min ;進樣量:20L;柱溫:30 ℃;檢測波長:280 nm。流動相條件見表1。
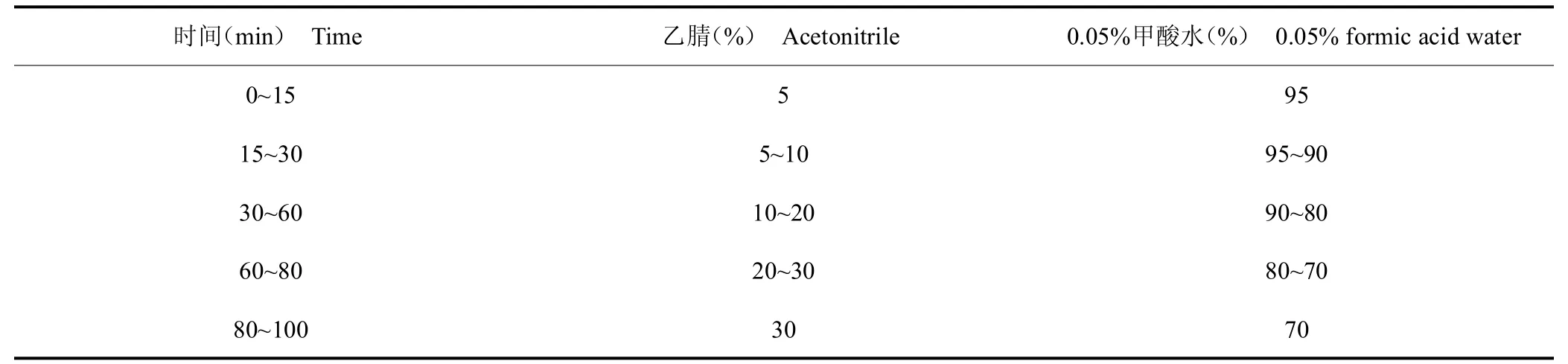
表1 流動相梯度洗脫表Tab.1 Gradient elution table
2.2 溶液的制備
2.2.1 供試品溶液的制備 取藥品膠囊內容物約3.0 g于具塞錐形瓶中,精密加入甲醇30 mL,超聲處理(功率150 W、頻率40 kHz)45 min,放冷,搖勻,過濾,蒸干,用甲醇定容至5 mL,過0.45m 濾膜,即得樣品溶液。
2.2.2 混和對照品溶液制備 精密稱取丹參素、原兒茶酸、原兒茶醛、迷迭香酸、黨參炔苷、紫草酸、丹酚酸B 對照品適量,加入甲醇溶解,搖勻,制備成含丹參素0.16 mg/mL、原兒茶酸0.20 mg/mL、原兒茶醛0.32 mg/mL、迷迭香酸0.25 mg/mL、黨參炔苷0.11 mg/mL、紫草酸0.13 mg/mL、丹酚酸B 0.19 mg/mL的混合對照品溶液,4 ℃下冷藏,備用。
3 方法學考察
3.1 精密度試驗
取同一批次止痛化癥膠囊(批號:160501),按照“2.2”項下方法制備供試品溶液,按照“2.1”項下色譜條件連續進樣6 次,記錄色譜圖。以重現性和分離度都較好的6 號峰原兒茶醛峰為參照峰S,計算各色譜峰的相對保留時間和相對峰面積。結果顯示,各特征峰相對保留時間RSD 為0.09%~0.40%、相對峰面積RSD為0.76%~2.94%,表明方法精密度良好。
3.2 重現性試驗
取同一批次止痛化癥膠囊(批號160501)5 份,按照“2.2”項下平行制備5 份供試品溶液,按照“2.1”項下色譜條件進樣,記錄色譜圖。以重現性和分離度都較好的6 號峰原兒茶醛峰為參照峰S,計算各色譜峰的相對保留時間和相對峰面積,結果顯示,各特征峰相對保留時間RSD 為0.08%~0.95%,相對峰面積RSD 為0.10%~2.87%,表明方法重復性良好。
3.3 穩定性試驗
取同一批次止痛化癥膠囊(批號160501),按照“2.2”項下方法制備供試品溶液,按照“2.1”項下色譜條件,分別于0 h、2 h、4 h、8 h、12 h、24 h 進樣,記錄色譜圖。以重現性和分離度都較好的6 號峰原兒茶醛峰為參照峰S,計算各主要色譜峰相對峰面積和相對保留時間。結果顯示,各特征峰相對保留時間RSD 為0.09%~1.30%、相對峰面積RSD 為 0.89%~2.83%,表明方法在24 h 內穩定性良好。
3.4 止痛化癥膠囊HPLC 指紋圖譜的相似度評價
將10 批止痛化癥膠囊分別按照“2.2”項下方法制備樣品溶液,按照“2.1”項下色譜條件依次進樣測定。按照國家藥典委員會發布的“中藥色譜指紋圖譜相似度評價系統(2004A 版)”分別對得到的色譜圖進行指紋圖譜分析,多點校正后進行自動匹配(時間窗為0.15),提取吸收信號較強、峰形明顯、穩定性較好的32個共有色譜峰,以6 號峰原兒茶醛峰為參照峰S,將參照峰的保留時間和峰面積作為1,分別計算共有色譜峰的相對保留時間和相對峰面積。32 個共有色譜峰生成對照圖譜R 與混合對照品溶液對照中,1 號峰為丹參素,2 號峰為原兒茶酸,6 號峰為原兒茶醛(S),23號峰為迷迭香酸,24 號峰為黨參炔苷,25 號峰為紫草酸,28 號峰為丹酚酸B。將10 批止痛化癥膠囊樣品所得指紋圖譜分別導入相似度評價系統,計算得相似度分別為0.981、0.976、0.990、0.982、0.991、0.981、0.978、0.983、0.973、0.981。結果見表2、表3、圖1、圖2。

表2 共有指紋峰相對保留時間Tab.2 Relative retention time of common peaks

表3 共有指紋峰相對峰面積Tab.3 Relative peak area of common peaks
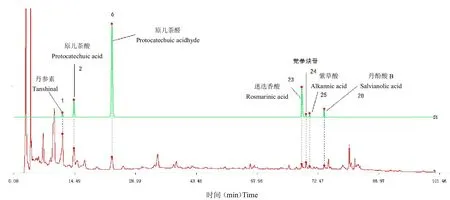
圖1 混合對照品(S1)和對照指紋圖譜(R)Fig.1 Mixed Control(S1)and Control Fingerprint(R)
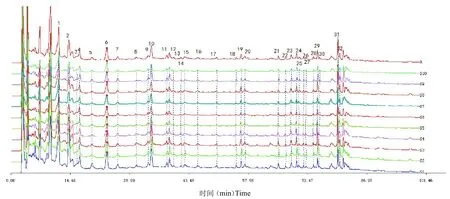
圖2 10批止痛化癥膠囊HPLC指紋圖譜Fig.2 10 Batches of Zhitonghuazheng capsule HPLC fingerprint
4 討論
4.1 供試品制備
本實驗采用50%、70%和100%甲醇分別對3 g 止痛化癥膠囊內容物進行超聲提取30 min,經考察,100%甲醇提取物基線平穩,響應值較高;以100%甲醇為提取溶劑,分別考察了超聲提取30 min、45 min 及60 min 的供試品制備條件,觀察其色譜峰的數目及其響應值,結果,45 min 和60 min 的色譜峰數目及響應值基本一致,表明45 min 超聲提取供試品基本完全。
4.2 波長的選擇
將供試品溶液進行190~400 nm紫外-可見光全波長掃描發現,在230 nm 和280 nm 處下各色譜峰的數目及豐度較好。經考察,230 nm的色譜峰基線不平穩、峰形差,280 nm的色譜峰基線平穩、分離度較好,適合指紋圖譜研究,故本研究選擇280 nm 為檢測波長。
4.3 流動相及流速的選擇
本實驗考查了甲醇-水、甲醇 0.05%甲酸水、乙腈-水、乙腈 0.25%醋酸水、乙腈 0.05%甲酸水等流動相系統,乙腈 0.05%甲酸水作為流動相梯度洗脫能夠使色譜峰分離度較好、基線平穩,所以選用乙腈 0.05%甲酸水流動相系統進行指紋圖譜的研究。在其他色譜條件一致的情況下,對流速條件進行了考察,分別設定流動相流速為0.5 mL/min、0.8 mL/min 及1 mL/min,經考察,流速為1 mL/min 的色譜圖整體分離效果較好,適合指紋圖譜研究。
4.4 進樣量的考察
4.5 柱溫的考察
在其他色譜條件一致的情況下,對其柱溫進行考察,分別設定柱溫30 ℃、35 ℃及40 ℃,并對其色譜圖進行分析,結果表明,柱溫30 ℃的色譜峰基線平穩、分離度較好、響應值較好,適合指紋圖譜研究。
5 止痛化癥膠囊HPLC指紋圖譜的首次建立
《中國藥典》2015 版一部規定,止痛化癥膠囊的質量利用薄層色譜法進行鑒別,采用高效液相色譜法(通則0512)測定丹參素的含量控制,本品每粒(0.3 g)含丹參以丹參素(C9H10O5)計,不得少于0.20 mg[9]。但這種單一成分的質量控制不足以體現藥品質量的優劣,中藥復方指紋圖譜是控制天然藥物質量的有效方式之一。查閱文獻,尚未發現止痛化癥膠囊指紋圖譜的研究報道,因此,本試驗借助HPLC技術彌補了止痛化癥膠囊指紋圖譜的空白,首次建立了不同批次止痛化癥膠囊的HPLC 特征指紋圖譜,且方法穩定、簡便、可靠。10 批止痛化癥膠囊樣品圖譜與生成的對照圖譜相匹配,相似度結果為0.981、0.976、0.990、0.982、0.991、0.981、0.978、0.983、0.973、0.981,均大于0.95 以上。可見,不同批次間止痛化癥膠囊的化學成分無較大差異、相似度較高、一致性良好,反映出止痛化癥膠囊的質量平穩。
6 結論
本試驗所建立的止痛化癥膠囊HPLC 指紋圖譜,分析方法簡單、準確、重復性好,共標定了32 個共有色譜峰;通過與對照品比對,指認出其中7 種成分,分別為丹參素、原兒茶酸、原兒茶醛、迷迭香酸、黨參炔苷、紫草酸、丹酚酸B。本研究可為止痛化癥膠囊的鑒別、質量控制提供一定的科學依據,指紋圖譜結合相似度分析可有效綜合評價不同批次止痛化癥膠囊的質量,進而對全面制定止痛化癥膠囊的藥品質量標準具有重要意義。
