手部骨纖維異樣增殖癥的診治
2019-03-07 06:05:42周強闞世廉張寶貴王曉剛
骨科臨床與研究雜志 2019年2期
關鍵詞:植骨
周強 闞世廉 張寶貴 王曉剛
骨纖維異樣增殖癥(fibrous dysplasia)是以骨的病理性纖維異樣增殖為基礎的良性病變。Lichetenstein[1]于1938年首先對該病作了描述,并將其命名為纖維異樣增殖癥。該病病因不確切,無遺傳性,臨床分為單骨型、多骨型及Albright綜合征。雖然該病臨床上可以發生于任何骨,但以脛骨、股骨、髂骨較多見,顱骨、肋骨次之。該病多為單骨型(75%~80%),多骨型較少見(20%~25%)[2],較少累及手部骨骼。通過檢索近年來國內外醫學文獻發現,單純發生于手部的骨纖維異樣增殖癥較少見,Hayter和Becton[3]、Gropper等[4]以及 Amillo等[5]分別于1984、1985和1996年報道了3例手骨單一掌骨纖維異樣增殖癥病例;吳繼明和王家瑚[6]與王明宇[7]于1990年和2012年分別報道2例手部骨纖維異樣增殖癥病例。本研究對6例手部掌、指骨纖維異樣增殖癥病例進行了總結。
資料與方法
一、資料
納入1980年3月至2016年1月天津市天津醫院收治的累及手部的骨纖維異樣增殖癥患者6例。其中男1例,女5例; 年齡6~27歲,平均15.7歲。病變分型包括單骨型4例,受累部位分別為掌骨1例、近節指骨2例和中節指骨1例,多骨型2例,受累部位分別為小指系列掌、指骨1例以及環、小指系列掌、指骨同時發病1例。其中3例患者發生病理骨折,骨折無明顯移位。
6例患者均表現為局部腫脹、輕度疼痛和手指活動輕度受限,但疼痛與手指活動無關,病變部位均有輕度壓痛。伴病理骨折患者有輕微外傷史,局部壓痛明顯。其余患者無外傷史。
術前X線示受累的掌骨和指骨囊性膨脹,骨質呈磨砂玻璃樣改變,病灶內部密度不均,可見不規則骨嵴影及點狀致密影。其中1例小指系列病變患者術前CT顯示第5掌骨及小指近、中、末節指骨存在囊性骨破壞,內部呈磨砂玻璃樣改變,夾雜小點狀鈣化斑。
2例手指系列病變患者術前實驗室檢查結果顯示血鈣、血磷及堿性磷酸酶水平均正常。
6例患者初步診斷均為良性腫瘤。4例術前考慮為骨纖維異樣增殖癥,2例考慮為內生軟骨瘤。
二、方法
在臂叢麻醉下行手指側方切口或手背側切口,暴露病變的指骨或掌骨。術中可見病變骨的骨皮質膨脹變薄,在皮質變薄的一側開窗,用刮匙刮除髓腔內腫物,為粉紅色砂礫狀顆粒。刮除徹底后用生理鹽水沖洗髓腔。沖洗干凈后于髓腔內填塞骨碎片。對其中4例患者行自體髂骨植骨,2例患者行同種異體骨植骨。縫合傷口。對刮除物行病理學檢查。
術后患手指石膏托固定3~4周。術后2周拆除逢合線。
術后隨訪30~56個月。以X線檢查骨愈合情況。末次隨訪采用手指總主動活動度(total active motion,TAM)系統評定方法對手功能和療效進行評價[8]。
結 果
術后病理學檢查結果示6例均為骨纖維異樣增殖癥,具體表現為正常骨結構消失,代之以增生的纖維母細胞和編織狀骨小梁,骨小梁排列紊亂,增生纖維母細胞呈長梭形,核仁長,染色深,染色質粗顆粒狀,膠原纖維呈束狀及漩渦狀排列。
6例患者術后均獲得隨訪。隨訪時間為30~56個月,平均41個月。6例患者傷口均愈合良好。接受異體骨植骨患者未出現異體組織排斥反應,病變部位外觀略顯隆起。復查X線示植骨與受區骨質愈合良好,隨時間推移,部分骨質塑性良好,髓腔部分形成。5例未見病變復發。1例掌骨病變病例術后22個月復發,再次行刮除植骨手術治療,再次手術后隨訪33個月未見復發。手TAM系統評定結果為優5例、良1例。
典型病例:患者女,17歲,因左手小指腫脹、輕度疼痛、活動受限2年7個月于2004年4月2日入院。無外傷史。體格檢查:左手小指輕度腫脹,小指及第5掌骨輕壓痛,小指伸直好,屈曲輕度受限。影像學檢查:X線示小指近、中、末節指骨及第5掌骨囊性膨脹,溶骨性破壞,病灶內密度不均,可見不規則骨嵴影及點狀致密影。CT示第5掌骨及小指近、中、末節指骨囊性骨破壞,內部呈磨砂玻璃樣改變,夾雜小點狀鈣化斑。術前實驗室檢查結果示血鈣、血磷及堿性磷酸酶水平均正常。初步診斷為良性腫瘤,主要考慮為內生軟骨瘤。于2004年4月8日行手術治療。術中在臂叢麻醉下行小指側方切口暴露患指第5掌骨及近、中、節末指骨,可見病變骨的骨皮質膨脹變薄,行皮質開窗,用刮匙刮除髓腔內腫物,內容為粉紅色砂礫狀顆粒。刮除徹底后用生理鹽水沖洗髓腔。采用同種異體骨進行植骨。縫合傷口。病理學診斷:骨纖維異樣增殖癥。術后2周拆除縫合線,未見感染及異體組織排斥反應。對患指行保護性石膏托固定4周。術后隨訪34個月,未見病變復發。患指TAM評定結果為良(圖1~4)。
討 論
自Lichetenstein[1]對纖維異樣增殖癥病進行描述后,該病有了明確的定義。該病病因目前尚不明確,可能與內分泌系統異常或骨發育異常有關。纖維異樣增殖癥是由成骨性間充質細胞活性受阻所致。纖維異樣增殖癥多是生長骨的病變,在臨床上患者出現癥狀的年齡差異很大,其中出現多骨病變者均為年輕患者。Bridge等[9]報道了57例纖維異樣增殖癥病例,其中單骨病變患者的平均發病年齡是14歲,多骨病變不合并內分泌紊亂患者的平均發病年齡是11歲,多骨病變合并內分泌紊亂患者的平均發病年齡是8歲。大多數纖維異樣增殖癥病例是孤立的單骨病變,少數為多骨病變,多骨病變合并內分泌紊亂的患者僅占約3%。單純發生在手部的纖維異樣增殖癥較少見,Hayter和Becton[3]、Gropper等[4]以及Amillo等[5]分別于1984、1985和1996年報道3例手部病例,均為累及掌骨的單骨型纖維異樣增殖癥。吳繼明和王家瑚[6]于1990年報道1例右手第3掌骨、第5掌骨及環、小指骨多發的纖維異樣增殖癥病例。王明宇[7]于2012年報道1例第5掌骨骨纖維異常增殖癥。我院在36年間僅收治6例纖維異樣增殖癥病例,約占同期手部骨腫瘤病例的0.16%。6例中單骨型4例,多骨型2例,其中2例多骨型病例均為手指系列掌、指骨同時發生纖維異樣增殖癥,國內外未見同類報道。系列掌、指骨發病的特點表明該病與發育異常有關。

圖1患者女,17歲,左手小指系列掌、指骨纖維異樣增殖癥。左手術前斜位和正位X線像圖2患者女,17歲,左手小指系列掌、指骨纖維異樣增殖癥。左手術前CT像圖3患者女,17歲,左手小指系列掌、指骨纖維異樣增殖癥。術中刮除的病理組織
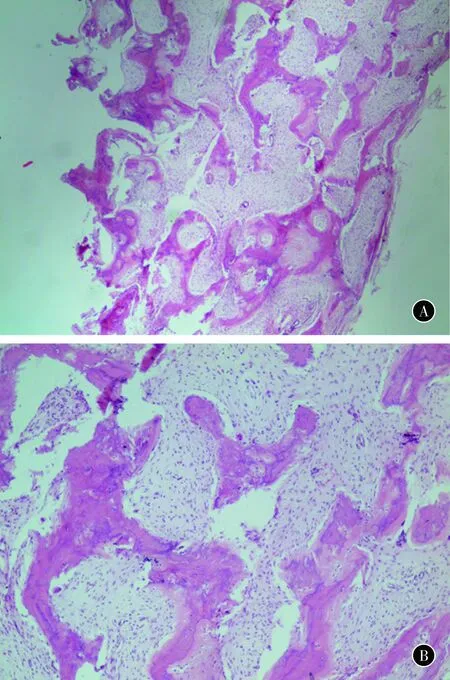
圖4患者女,17歲,左手小指系列掌、指骨纖維異樣增殖癥。刮除組織病理學檢查鏡下圖AHE×40BHE×100
多數纖維異樣增殖癥患者的主要臨床癥狀是輕微疼痛、腫脹以及局部壓痛,病理骨折是常見的并發癥。部分臨床癥狀不明顯的纖維異樣增殖癥患者因發生病理骨折而發現該病。本研究組6例患者中,3例因輕微外傷發生病理骨折而就診。由于受累骨的堅固程度受到明顯影響,肢體可出現彎曲畸形。顱骨及頜面骨受累的患者可出現視力下降或聽力受損,有時可出現面部畸形。纖維異樣增殖癥的癥狀較輕,病程較長,可長達數年或數十年之久,因此患者有時在青年或老年時期出現癥狀而被發現該病。而病變累及手部的患者,因病變部位相對表淺,且在手部使用過程中容易發現,就診較早,本研究組患者平均就診年齡為15.7歲。
Tarkkanen等[10]指出,骨纖維異常增殖癥是一種病因不明、進展緩慢的自限性良性骨纖維組織疾病。該病患者的正常骨組織被吸收,而代之以均質梭形細胞構成的纖維組織和發育不良的網狀骨骨小梁,可能系網狀骨未成熟期骨成熟停滯或構成骨的間質分化不良所致。骨纖維異常增殖癥在臨床中并非罕見,約占全部骨新生物的25%,占全部良性骨腫瘤的7%,其中單骨型約占70%,多骨型不伴內分泌紊亂者約占30%,多骨型伴內分泌紊亂者約占3%。
發生于手部的骨纖維異常增殖癥應與下列腫瘤鑒別診斷:(1)內生軟骨瘤,好發于手和足的短管狀骨,X線可見病變的透明區內有鈣化斑點。(2)動脈瘤樣骨囊腫,其臨床和X線表現與骨纖維異常增殖癥均有相似之處,但動脈瘤樣骨囊腫多為偏心性,具有中度侵蝕性,常可穿破骨皮質包殼,其邊緣輪廓模糊不清,呈蟲蛀狀,其骨皮質常膨脹如氣球狀,穿刺可見新鮮血液,穿刺時常有血液搏動感。(3)骨巨細胞瘤,多見于20歲以上患者,好發于長骨的骨端,病變呈多房狀或泡沫狀,具有高度偏心性和膨脹性,有一定的侵蝕性,可穿透骨皮質累及骨骺。
發生于手部的纖維異樣增殖癥的治療方法為病灶刮除并植骨。術中須將病灶徹底刮除,不主張應用甲醛等固定液擦拭腔壁。植骨可采用自體髂骨或同種異體骨。病變復發率主要取決于術者對該病的認識及術中對病變的清除是否徹底。本研究組中1例患者在術后22個月出現病變復發,主要考慮為病灶刮除不徹底,后經再次手術刮除并植骨,再次手術后隨訪33個月未見復發。
猜你喜歡
實用手外科雜志(2022年2期)2022-08-31 09:48:22
現代儀器與醫療(2022年2期)2022-08-11 09:53:42
脊柱外科雜志(2021年6期)2021-12-07 06:16:07
貴州醫科大學學報(2016年8期)2016-10-12 05:35:46
中國衛生標準管理(2015年10期)2016-01-15 00:48:55
川北醫學院學報(2015年5期)2015-12-05 08:22:31
西南軍醫(2015年3期)2015-04-23 07:28:16
湖北科技學院學報(醫學版)(2015年3期)2015-02-28 19:43:55
中國中醫藥現代遠程教育(2014年15期)2014-03-01 04:27:59
河南醫學研究(2014年5期)2014-02-27 14:52:51
