常用藏藥材叢菔質量標準提升
2019-02-10 10:58:18
中國民族民間醫藥 2019年24期
1.青海省藏醫藥研究院,青海 西寧 810006;2.金訶藏藥股份有限公司,青海 西寧 810003
叢菔現行質量標準載于《中華人民共和國衛生部藥品標準 藏藥》第一冊[5]和《青海省藏藥炮制規范》2010年版[7],質量控制項目僅限于性狀和顯微鑒別,由于藏醫口耳相傳的傳統以及地域環境不同導致對叢菔藥材的來源尚不明確,筆者在文獻資料的基礎上結合藏區各地對叢菔藥材的使用情況,確定了叢菔的藥材來源,為十字花科植物寬果叢菔Solms-laubachiapulcherrima(Maxim.) Bofsch.的干燥全草,并對其化學成分定性鑒別、水分和重金屬控制及含量測定方法進行了詳細研究,為更好地控制叢菔藥材的質量提供了依據。
1 儀器與試藥
1.1 儀器 超聲波清洗器(KQ-250型);高效液相色譜儀(島津LC-20AT); METTLER AG204萬分之一分析天平,METTLER AX205十萬分之一分析天平(瑞士梅特勒); HH-4恒溫水浴鍋(上海科學儀器廠);硅膠G薄層板(青島海洋化工廠);TAS-986原子吸收分光光度計(北京普析通用儀器有限責任公司),GFH-986石墨爐電源,AFS-830原子熒光光度計(北京吉天儀器有限公司)。
1.2 試藥 迪馬C18色譜柱;β-谷甾醇對照品(批號:872-200204供含量測定用);精氨酸對照品(批號:872-201304供含量測定用);以上對照品購自中國藥品生物制品鑒定所;雙蒸餾水(自制);乙腈、甲醇為色譜純,其他試劑均為分析純。鉛標準單元素溶液:標準值為1000μg·mL-1,證書編號為GBW08619;砷標準單元素溶液:標準值為1000μg·mL-1,證書編號為GBW08611;汞標準單元素溶液:標準值為1000μg·mL-1,證書編號為GBW08617;叢菔對照藥材(溯源樣品,采自青海貴南):由青海藏醫學院尕瑪措尼教授鑒定為十字花科植物寬果叢菔Solms-laubachiapulcherrima(Maxim.) Bofsch.樣品信息詳見表1。

表1 叢菔藥材收集情況

2 方法與結果
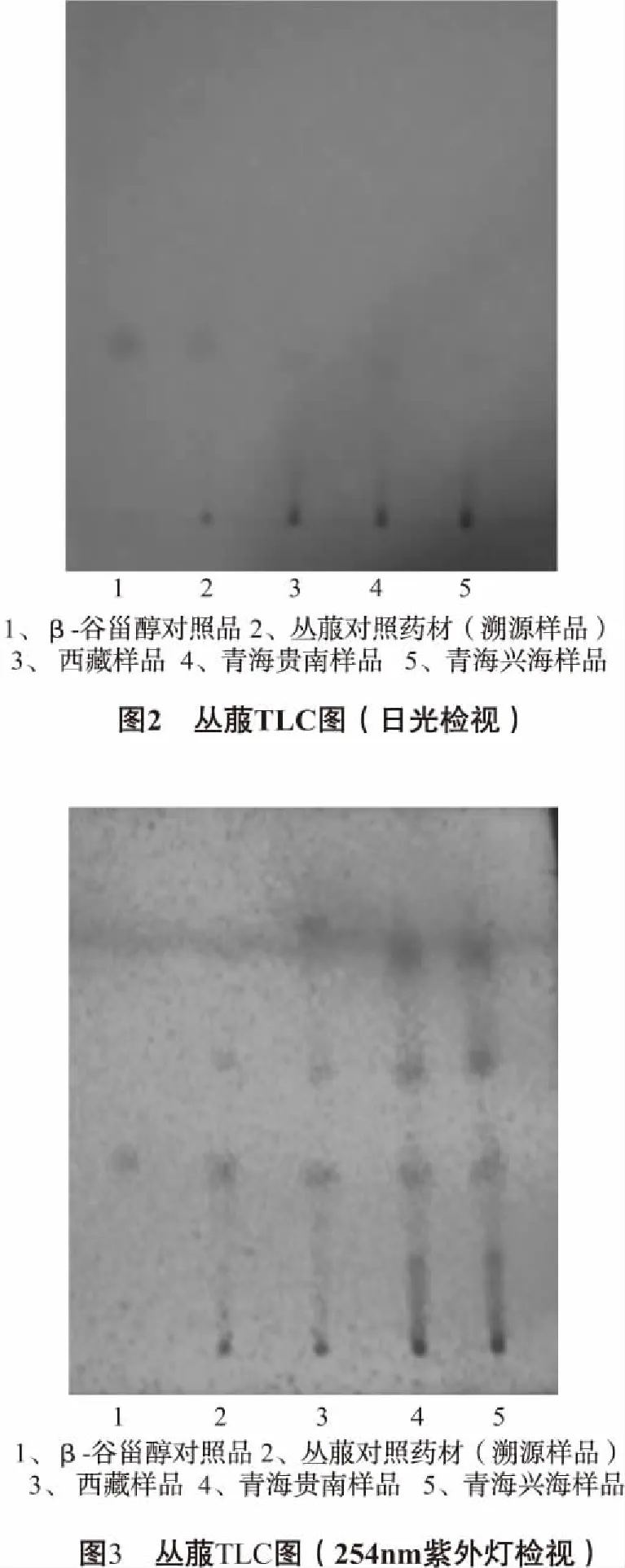
2.1 β-谷甾醇的TLC鑒別 取本品粉末2.0 g,加三氯甲烷30 mL,超聲處理(功率300 W,頻率40 khz)30 min,濾過,濾液濃縮至2 mL作為供試品溶液。另取叢菔對照藥材2.0 g,同法制成對照藥材溶液。再取β-谷甾醇對照品適量,加乙醇制成每1 mL含1 mg的溶液,作為對照品溶液。按TLC法試驗,吸取上述兩種溶液各10 μL,分別點于同一硅膠GF254薄層板上,以三氯甲烷-丙酮 (25∶1)為展開劑,展開,取出,晾干,噴以10%硫酸乙醇溶液,并在105 ℃加熱至斑點顯色清晰,置日光和紫外光燈(254 nm)下檢視,供試品色譜中,在與對照藥材色譜和對照品色譜相應的位置上,顯相同的紫紅色斑點和熒光斑點(如圖1和圖2所示)。
2.2 檢查
2.2.1 水分 取叢菔藥材粉末(過3號篩)約2 g,精密稱定,照水分測定法《中國藥典》2015年版一部附錄IXH第一法烘干法測定。每批平行測定2份,結果見表2。
2.2.1 重金屬檢測 隨機抽取9批樣本中3批原藥材,按照《中國藥典》2015年版一部附錄Ⅸ B項下(鉛、砷、汞)的測定方法進行測定,測定結果見表3。
2.3 浸出物測定 取叢菔藥材粉末(過3號篩)約2~4 g,精密稱定,以水為溶劑,照水溶性浸出物測定法熱浸法測定,每批平行測定2份,測定結果見表2。
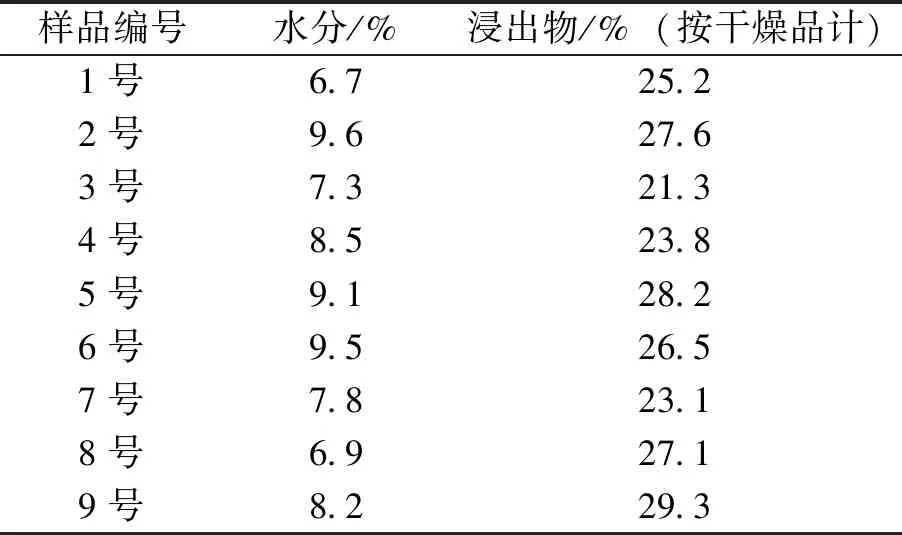
表2 叢菔藥材水分及浸出物測定結果

表3 叢菔藥材中重金屬測定結果
2.4 含量測定
2.4.1 色譜條件與系統適用性試驗 用十八烷基硅烷鍵合硅膠為填充劑;0.1 mol·L-1醋酸鈉緩沖液(pH=6)-乙腈(84∶16)為流動相;檢測波長為360 nm,柱溫30 ℃。理論板數按精氨酸峰計算應不低于2000。
2.4.2 對照品溶液的制備 精密稱取精氨酸對照品適量,加蒸餾水制成每1 mL含0.2 mg的溶液,即得精氨酸貯備液;精密吸取1.0 mL貯備液于10 mL量瓶中,加入0.5 mol·L的碳酸氫鈉溶液和1%DNFB乙腈溶液各1 mL,避光,于60 ℃水浴上反應30 min,取出,冷卻,以0.05 mol·L磷酸鹽緩沖溶液(pH=6.8)定容,即得20.0 μg·mL-1的對照品溶液。
2.4.3 供試品溶液的制備 取1 g蓯菔藥材粉末,精密稱定,置具塞錐形瓶中,精密加入20 mL水,稱定重量,超聲30 min,再稱定重量,用水補足減失的重量,搖勻,過濾,取2.0 mL續濾液于10 mL容量瓶中,加入0.5 mol·l-1碳酸氫鈉溶液和1%DNFB乙腈溶液各1 mL,避光,于60℃水浴上反應30 min,取出,冷卻,以磷酸鹽緩沖液定容,即得供試品溶液。[8]
2.4.4 提取方法考察 對樣品分別進行超聲處理、回流提取,結果超聲處理的效果最好,可消除對所有待測組分的干擾并且含量較高。結果見表4。
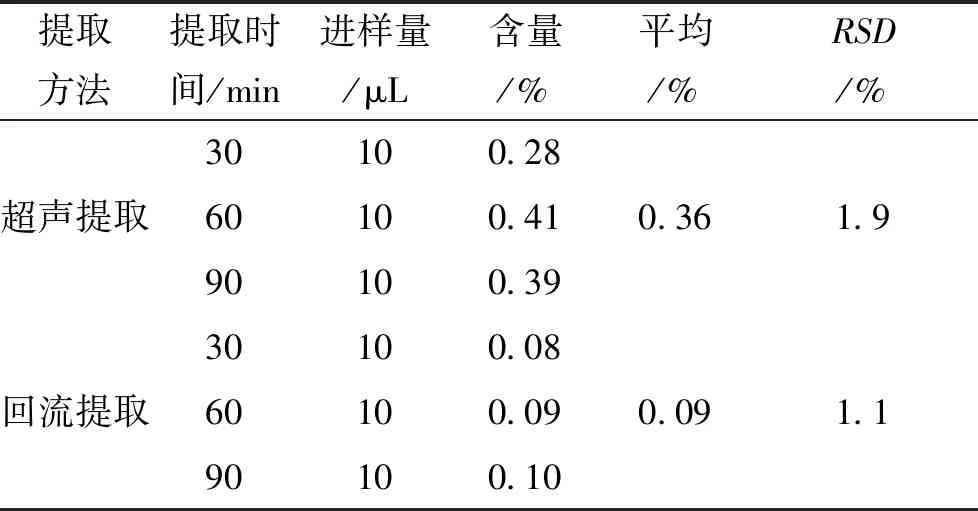
表4 提取方法學考察試驗結果
2.4.5 測定波長的選擇 取對照品溶液和供試品溶液分別在190~400 nm的波長內測吸光度,結果在360 nm處有最大吸收,故選此波長為測定波長。
2.4.6 線性關系的考察 精密吸取對照品溶液3、6、9、12、15 μL注入液相色譜儀測定,按2. 1項下的色譜條件測定峰面積,以進樣量為橫坐標(X),以峰面積的積分值為縱坐標(Y)繪制標準曲線。得精氨酸回歸方程為Y=229766X-41832.50,r=0.9992。結果表明,在上述色譜條件下,精氨酸在0.0612~0.306 μg范圍內呈良好的線性關系。結果見表5。

表5 線性關系實驗結果(精氨酸)
2.4.7 精密度試驗 精密吸取精氨酸對照品溶液10 μL,重復進樣6次,測得精氨酸峰面積RSD為1.4%。表明儀器進樣精密度良好。結果見表6。

表6 精密度試驗結果
2.4.8 穩定性試驗 精密吸取樣品溶液10 μL,分別于2,6,8,10,12 h進樣,測定精氨酸的含量,結果RSD為1.8%,即樣品溶液在12 h內穩定。結果見表7。
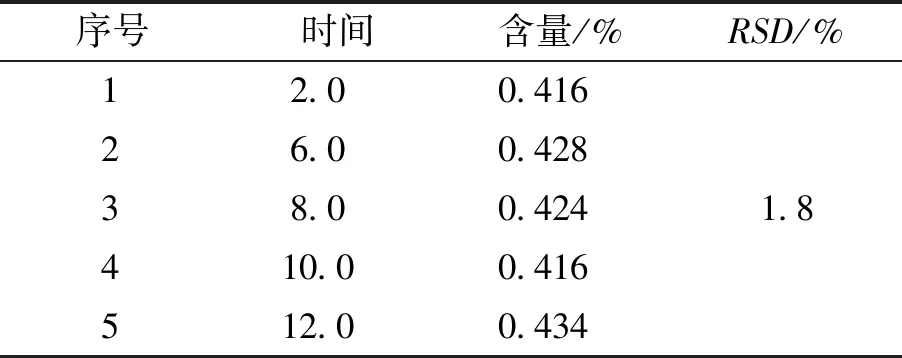
表7 穩定性試驗結果
2.4.9 重現性試驗 取不同批次樣品5份,按【含量測定】項下色譜條件測定,測定含量RSD為1.3%,表明該方法的重現性良好。結果見表8。
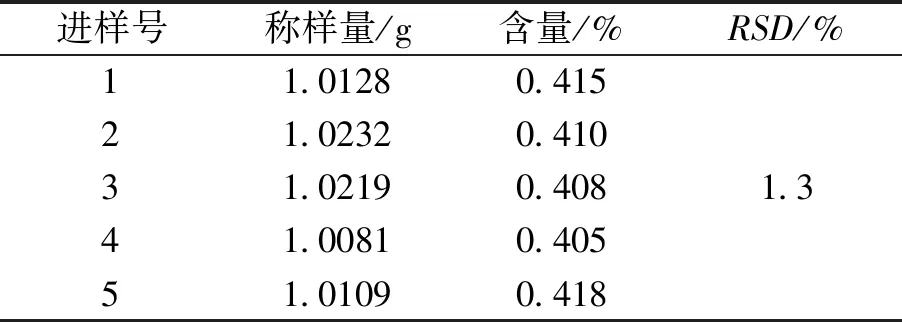
表8 重現性試驗結果
2.4.10 回收率試驗 取已知含量的樣品6份,精密加入精氨酸對照品溶液,按樣品制備方法和測試條件進行測定,計算得平均回收率為97.0%,RSD為0.9%。實驗結果見表9。

表9 加樣回收實驗結果
2.4 樣品測定 取1g樣品粉末,精密稱定,置具塞錐形瓶中,精密加入20 mL水,稱定重量,超聲30 min,再稱定重量,用水補足減失的重量,搖勻,過濾,取2.0 mL續濾液于10 mL容量瓶中,加入0.5mol·l-1碳酸氫鈉溶液和1%DNFB乙腈溶液各1 mL,避光,于60 ℃水浴上反應30 min,取出,冷卻,以磷酸鹽緩沖液定容,即得。吸取10 μL進樣,測定,結果3批樣品(2個平行樣)中精氨酸平均含量為0.314%。結果見表10。

表10 樣品中精氨酸的含量
3 討論
3.1 薄層色譜 研究過程中分別對叢菔所含不同成分(亞油酸、亞麻酸)的進行了薄層色譜分析,但始終效果不理想,只有β-谷甾醇的薄層色譜圖較為清晰,易于辨認,效果較好,通過對展開系統的篩選,分別以三氯甲烷-丙酮 (25∶1)、乙酸乙酯-丙酮 (20∶6) 、三氯甲烷-乙酸乙酯-丙酮 (12∶7∶3)等展開系統進行試驗,發現以三氯甲烷-丙酮 (25∶1)展開后效果最佳,Rf值符合要求;分別用10%硫酸乙醇溶液和香草醛硫酸溶液進行顯色實驗,發現10%硫酸乙醇溶液效果較好,斑點清晰,在日光下及紫外燈下均可見清晰斑點,說明本方法專屬性強、操作簡便,方法穩定。
3.2 水分 按中國藥典2010年版一部附錄IXH第一法烘干法測定9批樣品水分,根據測定結果顯示,暫定水分不得過12.0%。
3.3 重金屬 3批樣品檢測結果顯示重金屬含量非常小,未超限度。由于藥材中的重金屬與產地的自然生態環境土壤、水分及空氣中的重金屬含量有直接的關系,叢菔主要生長在海拔3500~4500 m的高山砂質土或巖石隙縫中,高海拔,重金屬無污染,控制藥材重金屬意義不大,故可暫列入標準正文。
3.4 浸出物 本研究依據藥典中關于浸出物的方法,分別對藥材進行了冷浸法和熱浸法的測定試驗,結果表明熱浸法優于冷浸法,且乙醇和水為溶劑的熱浸法比較顯示,以水為溶劑效果更好,因此選用以水為溶劑的熱浸法為浸出物測定方法。
3.5 含量測定 關于叢菔藥材的定量測定方面研究資料很少,巴桑旺姆等[8]曾用HPLC法測定叢菔藥材的精氨酸含量,筆者在上述方法的基礎上增加了樣品量,并對樣品制備方法和流動相進行了調整,使得精氨酸的分離度更好,準確度更高。鑒于考慮叢菔藥材中精氨酸的含量有所差異且9批樣品所得平均值后,故規定本品按干燥品計算,含精氨酸(C6H14N4O2)不得少于0.20%。
