超高效液相色譜-大氣壓化學電離-串聯質譜法測定烘焙咖啡中丙烯酰胺
2019-01-29 08:12:54朱銘立楊黎忠張衛鋒蔡成元周向東
色譜 2019年2期
關鍵詞:檢測
朱銘立, 楊黎忠, 張衛鋒, 蔡成元, 周向東
(1. 廣州市農產品質量安全監督所, 廣東 廣州 510308; 2. 珀金埃爾默企業管理(上海)有限公司, 上海 201203)
丙烯酰胺是一種白色晶體化學物質,其分子式為CH2CHCONH2,易溶于水、乙醇、醚及三氯甲烷等[1]。丙烯酰胺屬于2A類致癌物,已有大量的動物模型實驗表明,丙烯酰胺具有神經、生殖生育和遺傳毒性[2]。2002年瑞典科學家[3]首次發現經高溫加熱的含淀粉食物會產生丙烯酰胺,進一步研究表明,丙烯酰胺為食物加工過程中發生“美拉德反應”時的副產物。2017年11月20日,歐盟[4]發布指令EU 2017/2158(已于2018年4月11日實施),制定了食品中的丙烯酰胺的基準水平和緩解措施,以減少食品中的丙烯酰胺的含量。在該指令中,規定了丙烯酰胺的基準水平:焙烤咖啡為400 μg/kg;速溶咖啡為850 μg/kg;咖啡替代產品為500~4 000 μg/kg。
目前,食品中丙烯酰胺的分析方法主要有氣相色譜(GC)法[5]、高效液相色譜(HPLC)法[6,7]、氣相色譜-質譜(GC-MS)法[8-10]和液相色譜-串聯質譜(LC-MS/MS)法[11-16]等。前兩種方法受雜質干擾大,靈敏度低,特異性差。而GC-MS法前處理需衍生化反應,操作步驟繁瑣,且耗時長,無法滿足大批量樣品的分析[8-10]。LC-MS/MS無需衍生化處理,操作簡便,且靈敏度高,近年來被廣泛應用于丙烯酰胺的檢測[11-16]。
在食品安全檢測方法中常用的大氣壓質譜離子源有電噴霧離子化技術和大氣壓化學電離技術。由于兩者離子化的方式不同,相對大氣壓化學電離(APCI)技術而言,電噴霧離子化(ESI)技術的基質效應影響更為顯著,導致方法的靈敏度降低,影響實驗方法的準確性[17,18]。目前LC-MS/MS法主要采用電噴霧離子化(ESI)技術對丙烯酰胺進行測定[11-16],鮮少有大氣壓化學電離技術應用于丙烯酰胺的測定。國內針對食品中丙烯酰胺的檢測方法為GB 5009.204-2014,該方法適用于熱加工(如煎、炙烤、焙烤等)食品中丙烯酰胺的測定,規定了液相色譜-大氣壓電噴霧-串聯質譜法和氣相色譜-質譜法用于測定食品中丙烯酰胺。該方法在前處理提取過程中采用水作為提取劑,而對于咖啡類樣品,水與咖啡粉末形成懸濁液體,難于通過離心獲得上清液,嚴重影響凈化步驟的進行。其次烘焙咖啡的基質非常復雜,采用電噴霧離子化技術測定丙烯酰胺存在嚴重的基質效應,很難對丙烯酰胺進行準確的定性定量檢測。
本文采用甲醇作為提取劑,避免了懸濁液的產生,然后通過HLB SPE小柱凈化,采用超高效液相色譜-大氣壓化學電離源-串聯質譜(UHPLC-APCI-MS/MS)法進行測定,同位素內標法定量,有效降低了基質干擾,操作步驟簡單,為咖啡類產品中丙烯酰胺的檢測提供了更為簡便高效的方法。
1 實驗部分
1.1 儀器、試劑與材料
QSight220超高效液相色譜-三重四極桿質譜儀(美國PerkinElmer公司), BP211D感量為0.01 mg的電子天平(德國Sartorius公司), N-EVAP氮吹儀(美國Organomation Associates公司), SI-T256旋渦混合器(美國Scientific Industries公司), Visiprep24TMDL固相萃取儀(美國Supelco公司), Milli-Q超純水機(美國Millipore公司), HLB SPE小柱(30 mg/1 mL,美國Waters公司)。
丙烯酰胺標準品(純度≥99%)、13C3-丙烯酰胺標準品(純度≥98%)、甲酸(色譜純)、甲醇(色譜純)和乙酸銨(色譜純)均購自美國Sigma-Aldrich公司;實驗用水由Milli-Q超純水機系統制得。實驗樣品購自當地超市。
1.2 溶液配制
分別準確稱取適量丙烯酰胺和13C3-丙烯酰胺標準品,用超純水溶解并定容,配成質量濃度為100 mg/L的標準儲備液,于3 ℃避光保存;移取適量標準儲備液,根據實驗需要用水逐級稀釋,并配制適當濃度的標準工作液,現用現配。
1.3 樣品前處理
準確稱取0.50 g混勻的試樣,置于10 mL具塞塑料離心管中,加入25 μL 2 mg/L13C3-丙烯酰胺同位素內標工作溶液,渦旋靜置2 min,再加入5 mL甲醇,渦旋混勻10 min,以10 000 r/min的速度離心5 min。取2 mL上清液過0.22 μm有機相微孔過濾頭至干凈的試管中,準確移取1 mL上述濾液于試管中(帶準確刻度的試管),加入約200 μL超純水,置于40 ℃下氮吹至約0.1 mL,用超純水準確定容至1 mL,待凈化。
采用2 mL甲醇和2 mL水活化HLB SPE小柱,將上述待凈化液轉移至HLB SPE小柱中,使其自然下滴進行樣品凈化,棄去前0.5 mL過柱的凈化液(避免活化柱子里剩余的水,導致樣品被稀釋),收集0.5 mL后段的凈化液,過0.22 μm有機相濾膜,供LC-MS/MS測定。
1.4 LC-MS/MS條件
1.4.1色譜條件
色譜柱:Perkinelmer Brownlee validated AQ C18色譜柱(100 mm×2.1 mm, 3.0 μm);柱溫:30 ℃;進樣量:10 μL;流動相:A相為含0.1%(體積分數)甲酸的5 mmol/L乙酸銨水溶液,B相為甲醇;流速:0.4 mL/min;等度洗脫:A∶B=90∶10;分析時間為2 min。
1.4.2質譜條件
質譜離子源:APCI電離源;掃描方式:正離子掃描;監測模式:多反應監測(MRM); APCI源電暈針電流:3 μA;反吹干燥氣流速:5 L/min;霧化氣流速:7.5 L/min;離子源溫度:200 ℃;熱表面誘導去溶劑(HSID)質譜接口溫度:200 ℃。其他質譜參數見表1。

表 1 MRM掃描模式下丙烯酰胺和13C3-丙烯酰胺的質譜參數
* Quantitative ion; RT: retention time; Q1: precursor ion; Q3: product ion; EV: entrance voltage; CE: collision energy.
2 結果與討論
2.1 提取溶劑的選擇
丙烯酰胺為強極性化合物,在水中的溶解度為204 g/100 mL(25 ℃),而在甲醇中溶解度為155 g/100 mL[19]。本文選擇了水、甲醇作為提取溶劑進行比較,采用水提取的溶液顏色明顯深于采用甲醇提取的溶液。而且水與咖啡粉末形成懸濁液,難于通過離心獲得上清液,嚴重影響凈化步驟的進行。分別對所得提取液進行了凈化后上機試驗,結果表明,采用水提取時,在目標化合物峰附近存在雜質峰,無法與目標化合物分離,嚴重干擾后面的色譜-質譜檢測。而甲醇提取溶液無明顯雜質干擾,因此最終選擇了甲醇作為提取溶劑。
2.2 凈化條件的優化
烘焙咖啡制品的基質較為復雜,含有色素、脂肪、蛋白質以及糖類等化合物,導致在使用液相色譜-質譜分析時,存在嚴重的干擾,需對提取液進行凈化處理。為了能夠達到凈化樣品、改善檢測效果的目的,美國食品藥品監督管理局(FDA)[14]發布的采用HPLC-MS/MS檢測咖啡中的丙烯酰胺方法和GB 5009.204-2014均使用Oasis HLB和Bond Elut-Accucat固相萃取柱聯用法對樣品進行凈化,但是聯用兩種固相萃取柱成本較高,并易使回收率降低。本研究采用HLB SPE小柱對提取樣品進行凈化處理,SPE小柱經甲醇和水活化后,將待凈化液轉移至HLB SPE小柱,使其自然下滴。因活化HLB SPE小柱時,殘余水分殘留在HLB SPE小柱內,為避免樣品凈化液被殘余水分稀釋,棄去前段0.5 mL凈化液,收集后段凈化液。采用HLB SPE柱凈化,可去除脂肪和蛋白質等雜質,防止色譜柱受到污染,確保實驗長期的穩定性。
2.3 質譜條件的優化
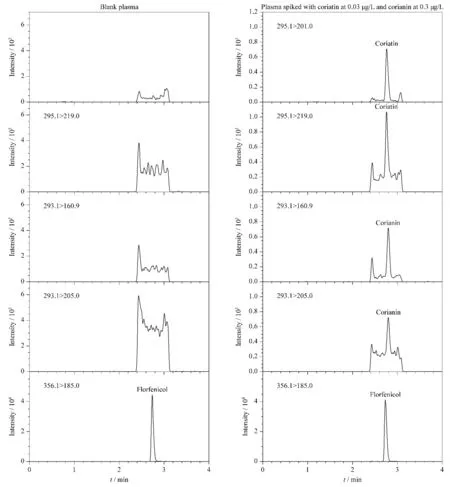
圖 1 在ESI源和APCI源下標準溶液和實際咖啡樣品中丙烯酰胺及13C3-丙烯酰胺的MRM色譜圖Fig. 1 MRM chromatograms of acrylamide and 13C3-acrylamide in standard solution and real coffee samples in ESI and atmospheric pressure chemical ionization (APCI) modes
丙烯酰胺通常采用電噴霧離子源(ESI)進行檢測分析[11-16],但在預實驗中發現,咖啡樣品基質在電噴霧離子源下檢測丙烯酰胺,產生嚴重的基質效應,對實驗結果帶來很大影響。在液相色譜-質譜聯用系統中,常見的大氣壓電離源有ESI源和APCI源。因離子化機理不同,較ESI源而言,APCI源的基質效應影響相對較小些[17,18]。本文采用1 μg/L丙烯酰胺標準溶液和實際樣品提取液(13C3-丙烯酰胺質量濃度均為10 μg/L)在ESI和APCI兩種電離模式下進行比較,以13C3-丙烯酰胺的峰面積響應值作為判斷依據。如圖1所示,在標準溶液中,ESI模式的靈敏度要優于APCI模式;在實際樣品中,ESI模式下存在嚴重的基質干擾,其靈敏度明顯低于APCI模式。從表2中可知,在ESI離子源下,實際樣品的基質效應為-99%,存在嚴重的基質效應,從而導致目標化合物響應值降低。而在APCI離子源下,樣品的基質效應為-20%,有較小的基質效應,目標化合物獲得良好的響應。因此相對ESI源而言,APCI源能大大降低咖啡的復雜基質帶來的嚴重基質效應問題,在實際樣品檢測中,靈敏度要優于ESI模式,更適合于咖啡中丙烯酰胺的檢測分析,因此最終選擇APCI模式用于咖啡中丙烯酰胺的測定。
2.4 線性范圍和檢出限
取丙烯酰胺質量濃度為0.5、1.0、2.0、5.0、10.0、50.0和100.0 μg/L的系列標準溶液(同位素內標13C3-丙烯酰胺質量濃度為10 μg/L)進行測定,以丙烯酰胺與同位素內標色譜峰峰面積比值為縱坐標y,以系列標準溶液的質量濃度(μg/L)為橫坐標x,繪制丙烯酰胺的標準曲線,其線性方程為y=0.116 2x+0.012 89。在0.5~100 μg/L范圍內,丙烯酰胺的質量濃度與對應的峰面積比值呈現良好的線性關系,相關系數r2為0.999。采用低濃度丙烯酰胺標準溶液,以超純水逐級稀釋并測定,分別以3倍和10倍信噪比在扣除基質效應的情況下確定方法的檢出限和定量限,本方法的檢出限為5 μg/kg,定量限為10 μg/kg。該方法檢測丙烯酰胺的線性范圍寬,靈敏度高,滿足咖啡類樣品中丙烯酰胺的日常的分析要求。
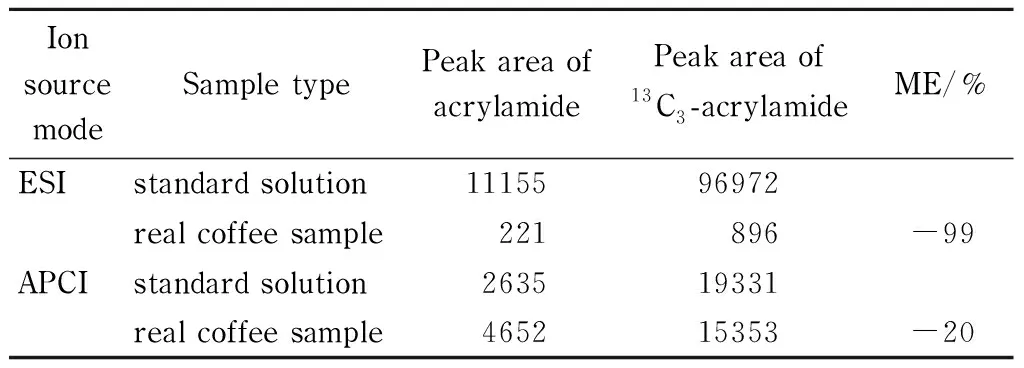
表 2 在ESI和APCI源下丙烯酰胺和13C3-丙烯酰胺在實際咖啡樣品中的基質效應
ME: calculation of peak area response ratio of13C3- acrylamide in real coffee sample and standard solution.
2.5 回收率和精密度試驗
取咖啡樣品,按照上述方法處理并測定,得到實際樣品中丙烯酰胺的含量為25.5 μg/kg。在上述樣品中添加丙烯酰胺標準溶液,3個加標水平為100、200和1 000 μg/kg,每個水平各添加6份樣品,并對樣品進行測定。如表3所示,丙烯酰胺的平均回收率為94.6%~115.0%,相對標準偏差(RSD)為2.8%~3.6%。該方法可獲得較滿意的回收率和精密度。
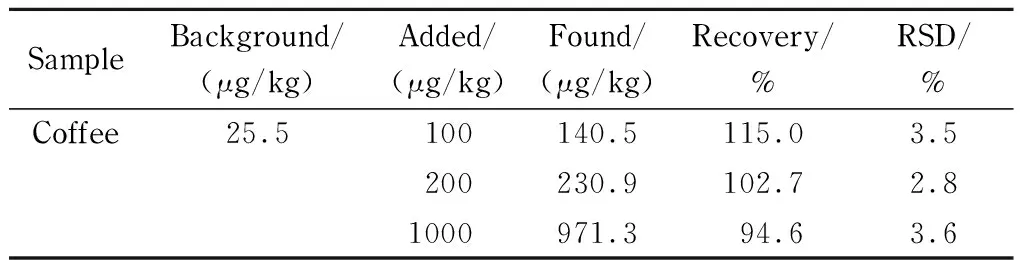
表 3 丙烯酰胺在咖啡樣品中的加標回收率及精密度(n=6)
2.6 實際樣品測定
采用本方法對市售12批次烘焙咖啡豆中丙烯酰胺的含量進行了分析。結果顯示,烘焙咖啡豆中均檢出丙烯酰胺,其含量均低于400 μg/kg,滿足歐盟發布的EU 2017/2158指令中規定烘焙咖啡豆中的丙烯酰胺的基準水平。
3 結論
本文建立了UHPLC-APCI-MS/MS測定食品中丙烯酰胺含量的方法。對比GB 5009.204-2014,本方法用甲醇提取,無需正己烷脫脂和HLB及Bond Elut-Accucat兩根固相萃取小柱凈化,簡化了前處理步驟,降低了檢測成本,同時利用APCI源測定咖啡類樣品中丙烯酰胺的含量,可大大降低咖啡的復雜基質帶來的嚴重基質效應問題。本方法靈敏度高,回收率良好,重現性優異,完全滿足咖啡中丙烯酰胺的日常檢測要求。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
