離子色譜-三重四極桿質譜聯用法測定奶粉中氟乙酸鈉
2019-01-22 11:53:36張秀堯蔡欣欣張曉藝李瑞芬
質譜學報 2019年1期
張秀堯,蔡欣欣,張曉藝,李瑞芬
(溫州市疾病預防控制中心,浙江 溫州 325001)
氟乙酸鈉是有機氟類殺鼠劑,屬A級劇毒物,大鼠口服半致死量為0.22 mg/(kg·bw), 人體口服半致死量為0.1~10 mg/(kg·bw)。由于氟乙酸鈉對人和動物的毒性極強、起效快、致死率高,并容易引起二次中毒,我國于1982年就明令禁止其生產、銷售和使用[1],但在新西蘭、澳大利亞、以色列和美國仍允許使用[2]。
目前,氟乙酸鈉的檢測方法有氣相色譜-質譜聯用法[3]、離子色譜法[4]、液相色譜-串聯質譜法[1, 5-6]等。其中,氣相色譜-質譜聯用法需要將樣品衍生化后才能測定,操作較繁瑣;離子色譜法靈敏度低,定性能力較弱,易出現假陽性結果;采用液相色譜-串聯質譜法檢測時,由于氟乙酸鈉在反相色譜柱上保留較弱,易受基質成分干擾[1],若進行衍生化,產率受樣品基質影響,且操作復雜耗時[5],也有采用反相和強陰離子交換固相萃取柱(PAX)進行凈化的報道,但因柱容量有限,易被樣品基質飽和而影響氟乙酸鈉回收率[6]。奶粉中氟乙酸鈉的測定方法主要有液相色譜-串聯質譜法[7]和液相色譜-四極桿/靜電場軌道阱高分辨質譜法[8]。前者將奶粉樣品用丙酮溶液提取,再將提取液過自填的AG 1-X8強陰離子樹脂固相萃取柱,用0.2 mol/L HCl溶液洗脫,用3-硝基苯胺衍生氟乙酸鈉,衍生物經Oasis HLB固相萃取柱凈化,樣品前處理比較繁瑣。后者先用正己烷脫脂,再用6 mol/L硫酸調節pH<1,用乙腈超聲提取,NaCl和MgSO4鹽析,氮吹濃縮,雖然步驟簡化但凈化效果不佳。
本研究擬建立離子色譜-三重四極桿質譜聯用方法檢測奶粉中氟乙酸鈉。采用高氯酸沉淀去除樣品中蛋白和脂肪等雜質,叔丁基甲醚萃取凈化,借助于離子色譜分離,三重四極桿質譜檢測,穩定同位素內標法定量分析,希望為檢測奶粉中氟乙酸鈉提供新途徑。
1 實驗部分
1.1 儀器與設備
離子色譜系統:美國Thermo Scientific公司產品,由Dionex ICS-1100離子色譜儀、淋洗液自動發生器(RFC-30)、電解再生抑制器(ASRS 500)和自動進樣器(DV-AS)組成,由變色龍工作站(7.22版)控制;QTRAP 6500三重四極桿/復合線性離子阱串聯質譜儀:美國AB SCIEX公司產品,由Analyst 1.6.2軟件控制;Multi Reax數顯型多管旋渦混合器:德國Heidolph公司產品;3-30K高速冷凍離心機:德國Sigma公司產品;N-EVAP氮吹儀(24孔):美國Organomation公司產品;2510超聲波清洗機:美國Branson公司產品;Gradient A10 Mill-Q 超純水器:法國Millipore公司產品。
1.2 材料與試劑
叔丁基甲醚、丙酮:均為HPLC級,德國Merck公司產品;高氯酸:上海桃浦化工廠產品;親水性聚丙烯(GHP)濾膜針式過濾頭(直徑13 mm,孔徑0.2 μm):美國Pall公司產品;氟乙酸鈉標準物質(純度97%):德國Dr. Ehrenstorfer公司產品;13C2-氟乙酸鈉同位素標準物質:新西蘭BDG Synthesis公司產品。分別用水溶解氟乙酸鈉、13C2-氟乙酸鈉標準物質并稀釋,得到1.30 g/L氟乙酸鈉標準貯備溶液和130 mg/L13C2-氟乙酸鈉標準貯備溶液,使用時用水稀釋至合適濃度的標準工作溶液。
1.3 實驗條件
1.3.1樣品前處理 稱取0.50 g奶粉試樣于15 mL塑料具塞離心管中,加入100 μL 1.3 mg/L13C2-氟乙酸鈉同位素內標液和2.0 mL水,旋渦10 s,再加入4.0 mL 3%高氯酸,旋渦10 s,超聲提取5 min,于6 ℃以10 000 r/min離心5 min,取約4 mL(pH 0.5~1.0)中間清液于15 mL具塞離心管中。向其中加入5.0 mL叔丁基甲醚,于多管旋渦混合器中旋渦3 min,以10 000 r/min離心2 min,移取上清液,用5.0 mL叔丁基甲醚重復提取殘渣1次,合并2次提取液。向提取液中加入100 μL 1%氨水丙酮溶液,混勻,于45 ℃氮吹至近干,加入1.0 mL 0.1%氨水溶液,旋渦10 s,過0.2 μm有機濾膜,待測。
1.3.2色譜條件 IonPac AS 19 型色譜柱(2 mm×250 mm×7.5 μm),AG19保護柱(2 mm×50 mm);ASRS 500陰離子抑制器(2 mm,外接水模式);KOH淋洗液由RFC-30自動在線產生;梯度淋洗程序:0~7 min(5 mmol/L KOH),7~8 min(5~50 mmol/L KOH),8~14 min(50 mmol/L KOH),14~20 min(5 mmol/L KOH);淋洗液流速0.30 mL/min;柱溫30 ℃;進樣體積10 μL。
1.3.3質譜條件 電噴霧電離(ESI)負離子多反應監測模式;離子化電壓(IS)-4 500 V;離子源溫度650 ℃;氣簾氣壓強277 kPa;噴霧氣壓強345 kPa;輔助加熱氣壓強345 kPa;碰撞氣設為Medium;氟乙酸鈉定量離子對為m/z77.0>57.0,去簇電壓-40 V,碰撞能量-15 eV;13C2-氟乙酸鈉的定量離子對為m/z79.0>59.0,去簇電壓-40 V,碰撞能量-15 eV;質譜峰駐留時間均為1 000 ms。
進樣運行開始時,離子色譜柱流出液經六通切換閥切換至廢液,5.50 min切換至離子源,質譜同時采集數據,直到8.00 min六通切換閥又將柱流出液切換至廢液中。
2 結果與討論
2.1 質譜條件優化
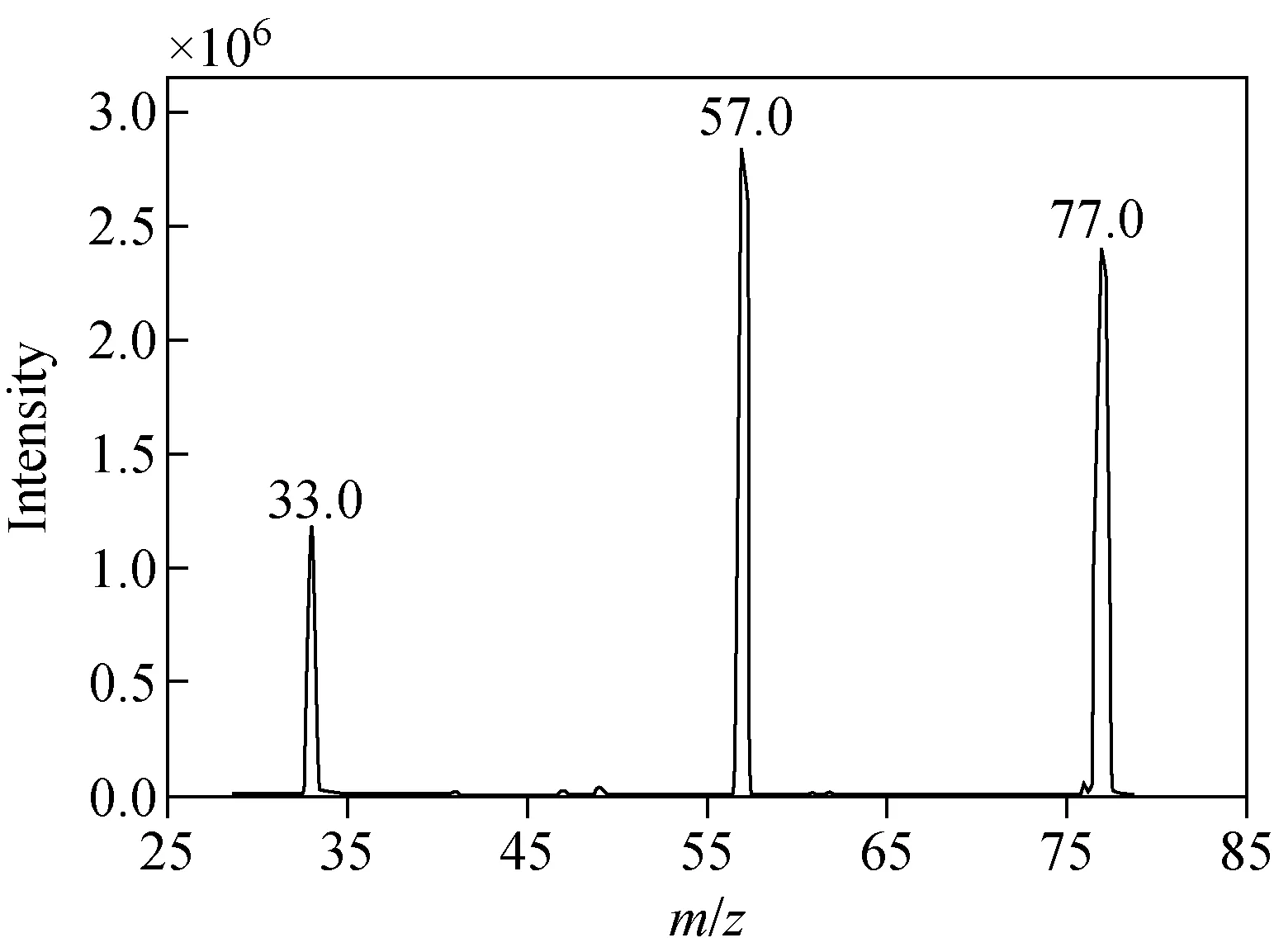
圖1 氟乙酸鈉子離子掃描譜圖Fig.1 Product ion scan spectrum of sodium monofluoroacetate
在ESI-模式下,優化檢測氟乙酸鈉(M)的質譜條件,Q1掃描時出現[M-Na]-峰,以[M-Na]-為母離子進行碰撞解離,通過子離子掃描,得到碎片離子m/z57.0[M-Na-HF]-和m/z33.0[M-Na-COO]-,示于圖1。然后,優化去簇電壓、碰撞能量等參數,使子離子的信號達到最強,優化后的結果顯示:m/z33.0碎片離子響應值較低,約為m/z57.0的10%,且基線較高,檢測實際樣品時干擾峰較多,因此,后續實驗僅選擇m/z77.0>57.0作為定量離子對。同時設定合適的峰駐留時間以確保色譜峰的采樣點數在15~20之間,以得到較好的定量重復性。優化后的質譜條件見1.3.3節。
2.2 離子色譜條件優化
氟乙酸鈉為弱酸鹽,lgKow為-3.89,水溶性強,使用常規反相色譜柱不能保留,文獻中有采用反相機理的Waters Aquity CSH Fluoro-Phenyl色譜柱進行分離[1],也有采用親水相互作用色譜(Hilic)機理的Waters Atlantis Hilic色譜柱進行分離[7],但氟乙酸鈉的保留均不理想,且易受樣品基質影響,保留時間不穩定[9]。離子色譜適用于氟乙酸鈉的分離[10-11],采用高容量AS19陰離子交換色譜柱,先用5 mmol/L KOH對樣品中氟乙酸鈉進行分離,然后用50 mmol/L KOH對色譜柱進行淋洗以去除強保留成分,色譜柱流出液經陰離子抑制器去除K+,流動相中KOH轉化成水,直接進入質譜系統進行檢測。采用0.5 mL帶濾頭的自動進樣瓶,進樣前先用1 mL水清洗進樣管路,進樣1 300 μg/L氟乙酸鈉標準溶液后,再進一針空白,結果表明,無氟乙酸鈉殘留。優化后的色譜條件見1.3.2節。
2.3 樣品前處理方法優化
2.3.1提取劑的選擇 氟乙酸為弱酸性水溶化合物,pKa 2.73[8],在pH<2的酸性條件下可被乙酸乙酯萃取[12-13]。奶粉樣品富含蛋白、乳糖、脂肪和礦物質等,選擇3%高氯酸作為提取劑對奶粉進行超聲提取,經低溫高速離心后取中間層提取液,可去除大部分蛋白和脂肪等雜質,此時提取液的pH值為0.5~1.0;此條件下氟乙酸呈分子狀態,易被有機溶劑萃取,在萃取液中加入100 μL 1%氨水丙酮溶液,使氟乙酸生成氟乙酸銨鹽,避免其在氮氣吹干過程中損失,再用0.1%氨水溶解殘渣,經0.2 μm濾膜濾除脂溶性雜質。稱取0.50 g奶粉樣品,加標10 ng氟乙酸鈉,測定氟乙酸鈉的響應值,再與0.50 g奶粉樣品處理液加標10 ng氟乙酸鈉的響應值進行比較,計算出高氯酸作為提取劑的提取率約為86%。
2.3.2萃取劑的選擇 稱取0.50 g奶粉樣品,加標10 ng氟乙酸鈉,分別用5.0 mL叔丁基甲醚和乙酸乙酯萃取。結果表明,叔丁基甲醚作為萃取劑的氟乙酸鈉響應值比乙酸乙酯作為萃取劑的響應值高20%,故選擇叔丁基甲醚作為萃取劑。
2.3.3提取液pH值的優化 取7份奶粉的高氯酸提取液,用高氯酸或氨水調pH值分別為0.5、1.0、1.5、2.0、3.0、4.0和5.0,再加入20 ng氟乙酸鈉標準溶液,用5.0 mL叔丁基甲醚萃取2次后測定,繪制pH值-氟乙酸鈉響應曲線,示于圖2。結果表明,在pH 0.5~1.0范圍內,氟乙酸鈉的響應值最高,故選擇提取液pH值范圍為0.5~1.0。
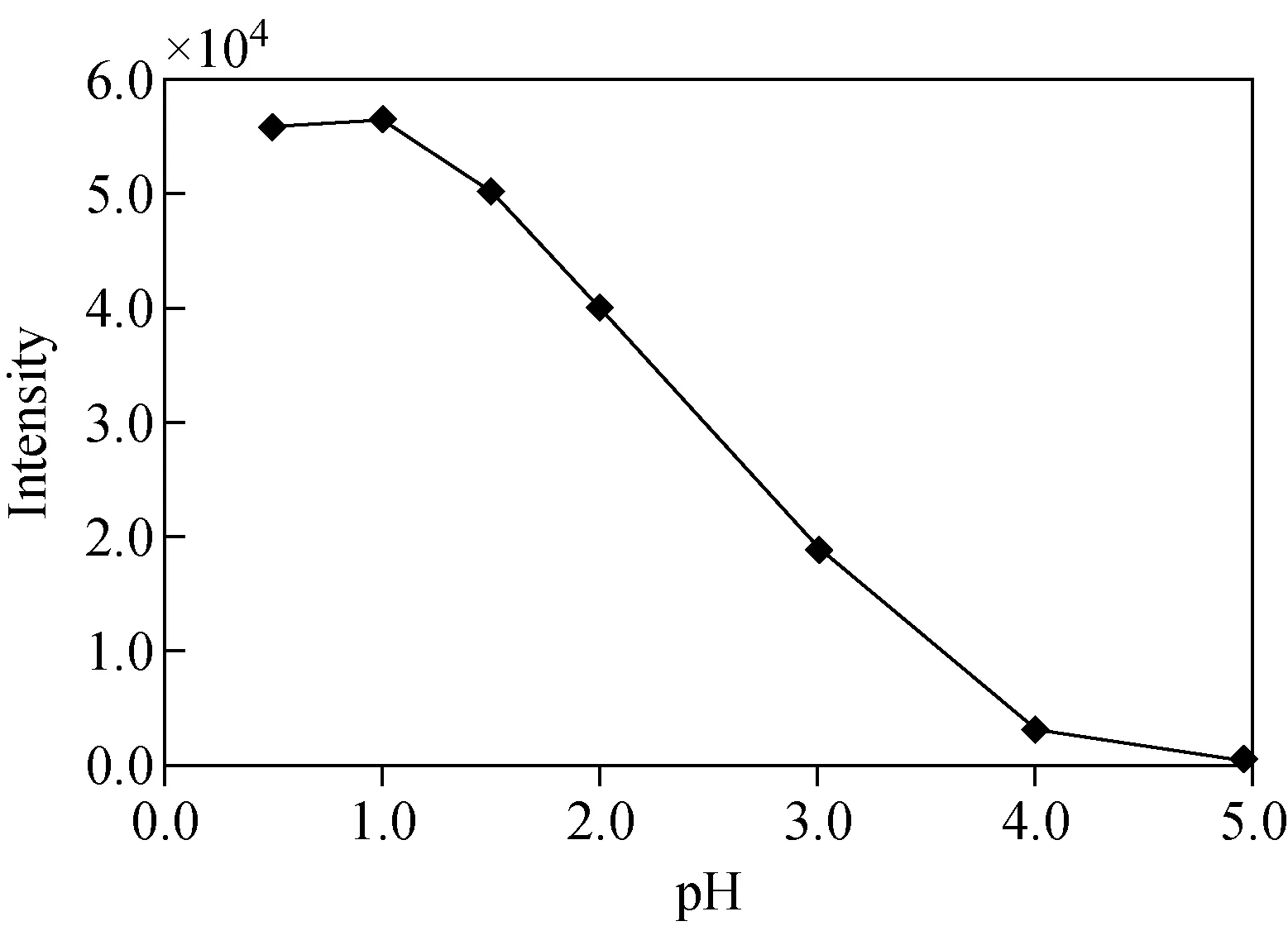
圖2 pH值對叔丁基甲醚萃取氟乙酸鈉的影響Fig.2 Effect of pH on sodium monofluoroacetate extracted by methyl tert-butyl ether
2.3.4萃取次數的選擇 將處于pH 0.5~1.0的高氯酸提取液加標10 ng氟乙酸鈉標準溶液,用5.0 mL叔丁基甲醚萃取3次,測定每次氟乙酸鈉的萃取量,通過除以3次萃取總量計算每次的萃取率。第1次叔丁基甲醚的萃取率約為60%,第2次約為30%,兩次的總萃取率約為90%,最終選擇采用5.0 mL叔丁基甲醚萃取2次。
2.3.5鹽析效應 本實驗考察了鹽析效應對氟乙酸鈉萃取率的影響,在高氯酸提取液中加標10 ng氟乙酸鈉標準溶液,再加入2.0 g氯化鈉后測定,并與不加氯化鈉的結果比較,發現鹽析效應對氟乙酸鈉的萃取率無明顯影響,故實際樣品萃取時無需加入氯化鈉進行鹽析。
2.3.6基質效應 本實驗采用穩定同位素內標法進行定量分析,可有效校正樣品中的基質效應。取空白嬰幼兒配方奶粉得到空白處理液,加入氟乙酸鈉標準溶液制成基質標準溶液(10 μg/L),與溶劑標準溶液(10 μg/L)相比評估基質效應[14],嬰幼兒配方奶粉中氟乙酸鈉的基質抑制效應約為33%。
2.4 方法的線性范圍、檢出限和定量限
將氟乙酸鈉標準溶液稀釋成濃度分別為0.3、1.0、5.0、50.0、100、600、1 300 μg/L的系列標準溶液(含130 μg/L13C2-氟乙酸鈉同位素標準溶液),采用MultiQuant定量軟件進行數據處理,以定量離子對與內標物的峰面積比值(y)對標準系列濃度(x,μg/L)進行線性回歸(權重取1/x),回歸方程為y=0.016 7x+0.002 55,相關系數為0.999 8,線性關系良好、線性范圍較寬。
向空白奶粉中加入低濃度氟乙酸鈉溶液,以分子離子對信噪比S/N≥3的樣品濃度作為檢出限(LOD),以S/N≥10的樣品濃度作為定量限(LOQ),測得方法的檢出限和定量限分別為0.2、0.6 μg/kg,測定上限為2.6 mg/kg。空白和加標氟乙酸鈉嬰幼兒配方奶粉的IC-MS/MS色譜圖示于圖3。
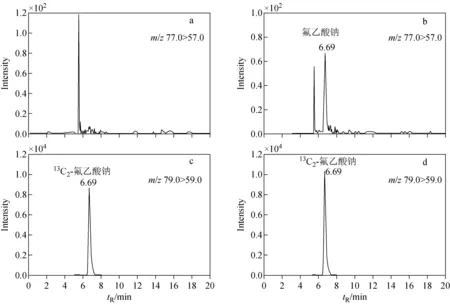
圖3 空白嬰幼兒配方奶粉(a,c)和0.6 μg/kg加標水平嬰幼兒配方奶粉加標樣品(b,d)的IC-MS/MS色譜圖Fig.3 IC-MS/MS chromatograms of blank infant formula (a,c) and infant formula added with sodium monofluoroacetate at 0.6 μg/kg (b,d)
2.5 加標回收率和精密度
向空白嬰兒配方奶粉和全脂奶粉樣品中添加不同濃度的氟乙酸鈉標準溶液,混勻,放置1 h,使待測成分與樣品基體成分相互作用達到平衡,再按1.3.1節方法處理樣品,其回收率和精密度結果列于表1。結果表明:氟乙酸鈉的加標回收率在89.7%~104%之間,相對標準偏差在0.50%~11%范圍內,符合痕量分析的要求。
2.6 實際樣品的測定
采用該方法檢測了5種進口嬰幼兒配方奶粉和3種全脂奶粉,均未檢出氟乙酸鈉。
3 結論
本研究采用離子色譜-三重四極桿質譜法測定奶粉中氟乙酸鈉。通過優化樣品的前處理過程,達到了較好的樣品凈化效果,降低了基質效應;通過優化離子色譜和質譜條件,得到了較好的分離效果和檢測靈敏度;采用穩定同位素稀釋內標法定量分析,結果準確、重現性好。本方法操作簡單、靈敏度高、線性范圍寬,可用于奶粉中氟乙酸鈉的日常檢測,也可應用于相似基質食品中氟乙酸鈉的測定。
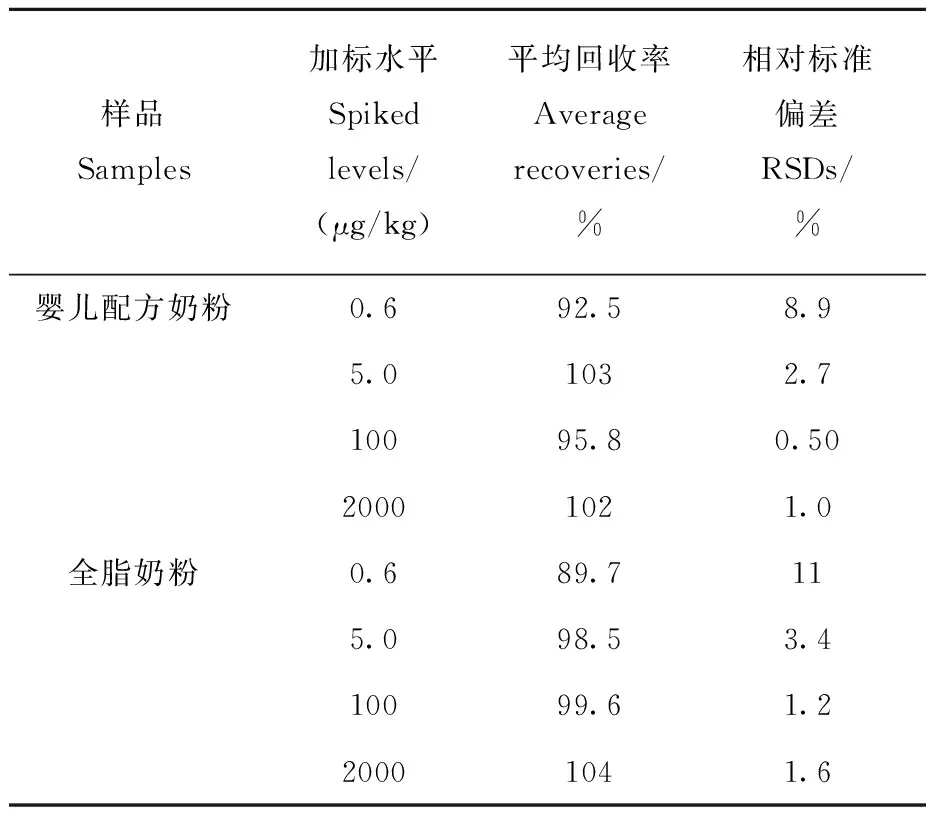
表1 在不同添加水平下,奶粉中氟乙酸鈉的回收率和精密度(n=6)Table 1 Recoveries and RSDs of sodium monofluoroacetate at different spiked levels in dairy powders samples (n=6)
