改性氧化石墨烯/聚氨酯復合包覆層的制備與性能測試
2019-01-19 07:45:20趙劉明李萬輝李梟銘王慶華郭雙鋒
火炸藥學報 2018年6期
關鍵詞:改性
趙劉明,陳 騰,李萬輝,李梟銘,李 強,王慶華,郭雙鋒,姜 煒
(1.南京理工大學國家特種超細粉體工程技術研究中心,江蘇 南京 210094;2.山西北方興安化學工業有限公司,山西 太原 030008;3.山西江陽化工有限公司軍代室,山西 太原 030008;4.西安近代化學研究所,陜西 西安 710065)
引 言
聚氨酯是聚氨基甲酸酯的簡稱,由含有多異氰酸酯基的化合物和含有多羥基的化合物聚合而成[1]。聚氨酯分子中含有酰胺基、酯基等基團,分子間容易形成氫鍵,有著較好的耐磨性和附著力;同時,聚氨酯具有較高的強度、高彈性和良好的低溫柔順性,這使得聚氨酯在包覆層領域應用廣泛[2-4]。
包覆層是固體推進劑裝藥的重要組成部分,是影響火箭發動機工作狀態和使用壽命的決定性因素。在實際應用過程中,聚氨酯的耐熱性較差,需添加填料以提高聚氨酯的熱穩定性及耐燒蝕性能。所選填料通常為碳酸鈣、炭粉等[5]無機顆粒狀填料,填料的添加增加了殘渣率,可能導致過濾器堵塞,無法正常工作。因此,需要對現有的聚氨酯包覆層配方體系進行改進,在提高耐燒蝕性能的同時,降低殘渣率。
石墨烯具有極佳的機械性能、導電性能和導熱性能[6-7]。石墨烯作為一種新型的納米材料,將其與高分子材料進行復合后,能夠提高高分子材料的耐燒蝕性[8-9]和力學性能[10-12]。石墨烯化學穩定性高,同時片層間的范德華作用力較強,易導致片層堆疊,難以均勻分散在聚合物基體中。為提高石墨烯在聚氨酯中的分散性,需要對其進行插層改性,減少團聚。
國外在研究聚氨酯包覆層領域起步較早,研究和發展歷史較長[13]。Reid等[14]采用原位合成的方法在含有預聚物羥基封端聚丁二烯的溶液中制得TiO2納米粒子,后澆鑄固化成聚氨酯復合材料,并研究了其作為固體推進劑包覆層的性能,發現相比于常規的粉末或溶液混合復合的方法,原位法制得的TiO2表現出了更高的燃燒催化劑活性;Lemos等[15]采用動態力學分析(DMA)研究了幾種含能增塑劑對復合推進劑包覆層玻璃化轉變溫度(Tg)的影響,結果表明1, 2-二(2-疊氮乙氧基)乙烷(BATEG)降低Tg的能力最強。國內吳淑新等[16]使用改性液體4, 4′-二苯基甲烷二異氰酸酯與端羥基碳氮雜環氯化聚醚多元醇為原料合成異氰脲酸酯改性聚氨酯彈性體,與普通聚氨酯相比,其熱穩定性得到有效提高;史愛娟等[3]采用端羥基聚醚預聚體和端異氰酸酯預聚體合成了聚氨酯包覆層,研究了異氰酸酯指數對包覆層力學性能、黏接性能和推進劑相容性的影響,發現黏接性能和拉伸強度隨R值增大而增大,相容性隨R值增大而變差。
本研究采用異佛爾酮二異氰酸酯改性氧化石墨烯,將其與原有聚氨酯包覆層體系復合,制得改性氧化石墨烯/聚氨酯包覆層材料,并對制得的材料進行性能研究。
1 實 驗
1.1 材料及儀器
聚氨酯包覆層A組分(羥基組分,含填料)、聚氨酯包覆層B組分(異氰酸酯組分,含填料),山西北方興安化學工業有限公司;氧化石墨,南京吉倉納米科技有限公司;異佛爾酮二異氰酸酯(IPDI),上海麥克林生化科技有限公司;乙酸正丁酯,成都市科龍化工試劑廠;N, N-二甲基甲酰胺(DMF)、正庚烷,國藥集團化學試劑有限公司。
D8 ADVANCE型X射線衍射儀,德國Bruker公司;Nicolet iS10傅里葉變換紅外光譜儀,美國Thermo Fisher Scientific公司;LabRAM ARAMIS型全自動激光拉曼光譜儀,法國HORIBA Jobin Yvon公司;AGS-500N臺式電子萬能試驗機,日本島津公司。
1.2 樣品制備
1.2.1 改性氧化石墨烯(MGO)的制備
稱量0.5g氧化石墨,加入至200mL DMF中,攪拌后超聲分散30min,得到氧化石墨烯分散液[17-18];將5g IPDI加入到上述分散液中,持續攪拌,在80℃、N2保護下反應24h;將所得的產物離心后,用乙酸正丁酯洗滌沉淀3次,以除去過量的IPDI,干燥后備用[19]。
1.2.2 MGO/聚氨酯(MGO/PU)復合包覆層的制備
將一定量的MGO加入到聚氨酯包覆層的A組分中,加入適量的乙酸正丁酯,超聲分散30min;50℃下保溫48h,以除去乙酸正丁酯;按照A組分和B組分質量比為1.0∶ 1.1稱取B組分,加入占A、B總質量10%的乙酸正丁酯降低黏度,并攪拌均勻;對上述混合液進行抽真空除泡處理,真空度為0.098MPa,保持5min后倒入模具中在45℃下保溫24h,制得MGO/PU包覆層材料。所制備試樣的MGO質量分數分別為2%、3%和5%。
1.3 測試方法
1.3.1 MGO的結構分析
采用X射線衍射儀進行XRD測試,采用銅靶,掃描角度5°~60°;采用紅外光譜儀進行紅外光譜測試,波長范圍4000~525cm-1;采用拉曼光譜儀進行拉曼光譜測試,測試波長為532nm,范圍600~2000cm-1。
1.3.2 復合包覆層的使用性能測試
拉伸試樣的制備按照GB/T 16421-1996標準,采用臺式電子萬能試驗機進行測試,測試溫度10℃,拉伸速率100mm/min;根據GJB 323A-96燒蝕材料燒蝕實驗方法,以氧-乙炔焰流為熱源,測定包覆層樣品的線燒蝕率;殘渣率測試在箱式爐中進行,測試條件:以5℃/min的速率升溫至800℃,保溫1h后,緩慢降至常溫,用試樣的剩余質量除以初始質量計算出殘渣率。
2 結果與討論
2.1 氧化石墨烯的改性效果
對改性前后氧化石墨烯粉末進行XRD表征,結果如圖1所示。
由圖1可知,GO在11.08°處有一尖銳的衍射峰,對應GO(0 0 1)晶面的特征衍射峰,MGO則在23.68°附近有一個較寬的衍射峰,表明改性過程中石墨的規整結構進一步被破壞,無序度增加[20],這有利于提高GO在有機溶劑中的分散性和與聚氨酯包覆層的相容性。
對MGO粉末進行紅外光譜測試,結果如圖2所示。
從圖2中可以看出,GO在3201cm-1處的寬峰為O-H的伸縮振動峰,表明GO含有大量的羥基官能團,異氰酸酯可通過與羥基的反應接枝到GO上;在1715、1613和1041cm-1處的峰分別為C=O、芳環骨架和C-O的伸縮振動峰。MGO在3320、2932和2856cm-1處的峰分別為N-H的伸縮振動峰和C-H的反對稱、對稱伸縮振動峰[21],O-H峰的消失和N-H、C-H峰的出現表明IPDI的異氰酸酯基與GO上的羥基反應較為徹底。在GO上接枝的IPDI提高了石墨烯與有機溶劑的相容性,有利于進一步與聚氨酯包覆層的復合。
對改性前后氧化石墨烯進行拉曼光譜表征,結果如圖3所示。
由圖3可知,GO和MGO均在1348、1590cm-1左右出現吸收峰,分別為石墨烯片層中無序或sp3雜化的碳原子振動的缺陷峰(D峰)和sp2雜化碳原子的面內伸縮振動峰(G峰)。D峰、G峰吸收強度之比(ID/IG)可以反映材料的石墨化程度[22],GO的ID/IG比值為0.89,經過IPDI改性后,ID/IG增至1.00,石墨化程度降低,表明IPDI改性劑的插層作用致使原有的石墨烯片層結構遭到進一步破壞,有利于提高GO和聚氨酯基體的相容性。
2.2 MGO的分散性及溶劑的選擇
聚氨酯包覆層A、B組分的黏度很高,流動性和流平性差,直接將A、B組分混合后攪拌,會產生大量氣泡[23]。經過抽真空除泡處理制得原始聚氨酯包覆層試樣,其截面和上表面形貌如圖4所示。
從圖4中可以看出,即使經過抽真空除泡,試樣內部和上表面仍零星分布有氣泡,這對包覆層的力學性能有較大影響。為減少制得試樣的氣泡含量,需要降低體系的初始黏度,從而增強抽真空除泡的效果,而使用溶劑是常見的黏度調節方法。
為更好地通過溶液共混法復合MGO與包覆層,對MGO在3種常見溶劑中的分散性進行了研究,GO和MGO的分散液照片如圖5所示。圖5(a)~(c)分別為GO在正庚烷、乙酸正丁酯和DMF中超聲分散30min后分散液的照片;圖5(d)~(f)分別為MGO在正庚烷、乙酸正丁酯和DMF中超聲分散30min后分散液的照片;分散液質量濃度均為1mg/mL。
由圖5可知,GO在DMF中的分散性較好,在乙酸正丁酯中分散性一般,在正庚烷中的分散性差,分散性與溶劑極性正相關[18]。MGO在DMF和乙酸正丁酯中分散性較好,在正庚烷中的分散性一般, MGO分散性仍與極性正相關,經過改性后的GO在有機溶劑中的分散性有明顯提高,其原因是經過IPDI改性后,MGO的極性增加,提高了其在極性溶劑中的分散能力,這與前述XRD的表征結果相一致。該包覆層材料要求固化溫度接近室溫,而DMF的揮發性較低,難以在較低溫度下完全烘干,同時聚氨酯固化反應有非常強的溶劑效應,DMF的高極性對聚氨酯固化反應不利[1];正庚烷揮發性高,但其極性較低對聚氨酯的溶解力弱,且對MGO的分散能力較弱;乙酸正丁酯揮發性較高,對聚氨酯的溶解能力較強[24],同時能夠較好地分散MGO。因此,選用乙酸正丁酯作為調節黏度的溶劑以制備MGO/PU復合包覆層材料。
MGO質量分數分別為0、2%、3%和5%時的聚氨酯包覆層試樣照片見圖6。
從圖6可以看出,選用乙酸正丁酯作為溶劑并結合真空除泡所制得的試樣具有平整光滑、無氣泡的表面。
2.3 復合包覆層的使用性能
MGO含量對聚氨酯包覆層定伸應力和延伸率的影響見表1和圖7。
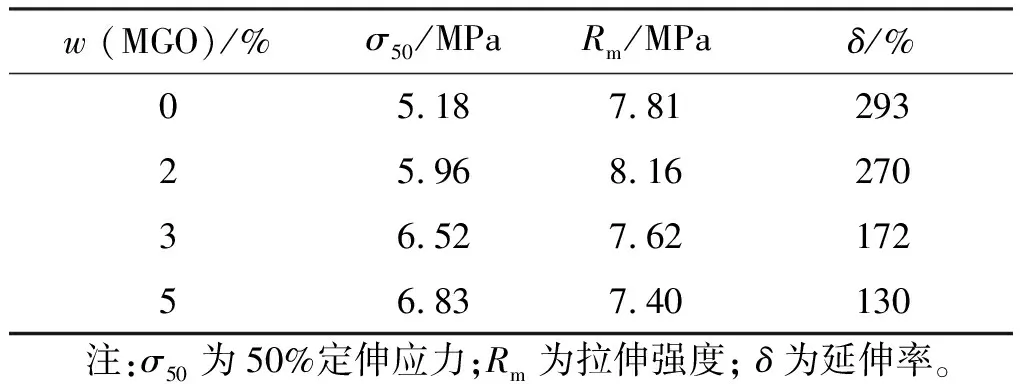
表1 不同MGO含量聚氨酯包覆層的定伸應力和延伸率Table 1 Stress and elongation of polyurethane coating layer
由表1和圖7可知,相較于未添加MGO的試樣,50%定伸應力提高31.85%,拉伸強度和延伸率分別下降5.25%和55.63%。隨著MGO含量的增加,聚氨酯包覆層的50%定伸應力呈現增大趨勢,拉伸強度先增大后減小,延伸率有明顯下降,脆性變大。其原因可能在于石墨烯添加量較多,片層間的范德華力使得石墨烯重新發生堆疊聚集,在拉伸過程中易產生應力集中,產生銀紋效應[25],致使試樣提前斷裂,導致延伸率和拉伸強度的下降。
采用氧-乙炔焰流為熱源,對包覆層試樣進行燒蝕性能測試,結果如圖8所示。
由圖8可知,隨著MGO的添加,線燒蝕速率(vL)有明顯的下降,但下降幅度逐漸變小。未添加MGO試樣的耐燒蝕性能較差,線燒蝕速率為0.654mm/s;當MGO質量分數達5%時,其線燒蝕速率為0.431mm/s,較未添加MGO時下降34.10%,耐燒蝕性能顯著提高。由于MGO具有較高的熱穩定性,提高了聚氨酯包覆層整體的熱穩定性[26],同時其能夠以片層結構分散于基體中形成曲折的通道,從而減緩燃燒過程中產生的氣體以及成炭的逸出[27],燃燒速率因此降低,提高了耐燒蝕性能。
使用箱式爐對包覆層試樣進行殘渣率測試,結果見表2。
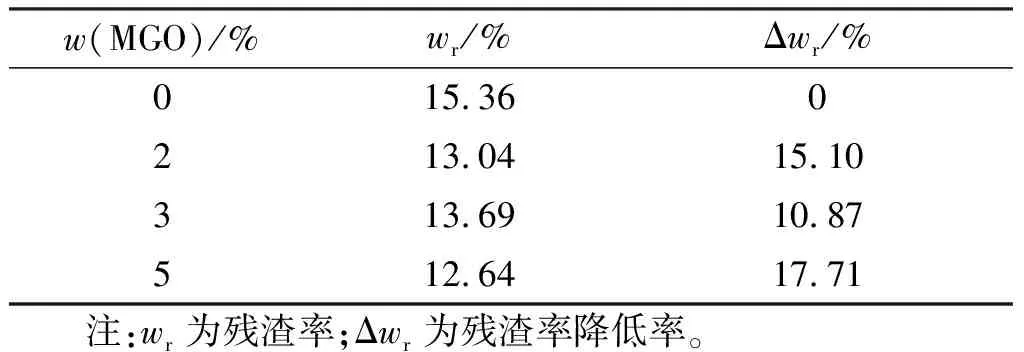
表2 不同MGO含量聚氨酯包覆層的殘渣率Table 2 Residue ratios of polyurethane coating layer with
從表2中可以看出,加入MGO后,殘渣率有所下降,但影響不顯著, 當MGO質量分數達到5%時,殘渣率為12.64%,相比于空白樣下降17.71%。分析認為,由于在原有體系中引入了可燃組分(MGO),原有包覆層體系中無機填料的相對含量降低;同時,MGO中擁有大量的含氧官能團,提高了O/C比值[2],可在包覆層熱分解過程中提供氧元素,減少因燃燒不充分導致的殘渣[28],從而降低殘渣率。
3 結 論
(1)經過IPDI改性后,IPDI成功插層到GO片層之間;MGO在有機溶劑中的分散性提高,在DMF和乙酸正丁酯中的分散效果較好,在正庚烷中的分散性一般。
(2)隨MGO添加量的增大,復合包覆層材料的50%定伸應力升高、拉伸強度先增大后減小,延伸率下降;MGO質量分數為5%時,其50%定伸應力為6.83MPa,拉伸強度為7.40MPa,延伸率為130%,相較于未添加MGO的試樣分別提升了31.85%,下降5.25%和55.63%。
(3)MGO的添加,提高了復合包覆層材料的耐燒蝕性能,并降低了殘渣率。MGO質量分數為5%時,線燒蝕速率降低34.10%,殘渣率降低17.71%。
猜你喜歡
紡織科學研究(2020年1期)2020-05-21 00:31:06
中國塑料(2016年12期)2016-06-15 20:30:07
中國塑料(2016年2期)2016-06-15 20:30:00
中國塑料(2016年2期)2016-06-15 20:29:59
中國塑料(2016年5期)2016-04-16 05:25:36
廣西林業科學(2016年3期)2016-03-16 05:43:30
中國塑料(2015年3期)2015-11-27 03:41:38
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
中國塑料(2015年4期)2015-10-14 01:09:19
