明日葉中4-羥基德里辛和黃色當歸醇的提取純化及鑒定
2019-01-11 02:13:32張連富
食品與生物技術學報 2018年11期
王 磊, 張連富
(江南大學 食品學院,江蘇 無錫214122)
明日葉(Angelica keiskei),是傘形科多年生草本植物,可藥食兩用[1]。嫩莖和葉可作為蔬菜用于炒、炸、涼拌或做湯,而全植物可入藥。研究表明,明日葉含有人體所需要的多種礦物質、氨基酸、維生素以及微量元素,是一種營養較全面、均衡的蔬菜[2]。此外明日葉還含有查爾酮類、香豆素類、類黃酮類、天然有機鍺等天然功能性成分[3],具有抗氧化、抗腫瘤、降血壓、降血脂、降血糖、改善睡眠、提高免疫力等保健功能[4]。均衡全面的營養以及優良的保健作用,賦予了明日葉很大的利用價值,以明日葉為原料開發出的產品也多種多樣,有保健茶[5]、保健酒[6]、固體飲料[7]、保健膠囊[8]、咀嚼片[9]、營養保健面條[10]、黑番茄醬[11]等。
其中 4-羥基德里辛(4-Hydroxyderricin,4-HD)和黃色當歸醇(Xanthoangelol,XAG)是明日葉中主要的功能性保健成分[12]。因此為了更好地開發和利用4-HD、XAG、明日葉及其相關的產品,需要對4-HD和 XAG進行深入的研究探討。然而,近年來對明日葉的研究主要集中在4-HD和XAG的功能保健作用上,明日葉中4-HD和XAG的提取工藝尚未見報道。因此本文以明日葉為原料,以4-HD和XAG的得率為評價指標,研究提取工藝參數(提取溶劑、溫度、時間、次數以及料液比)對提取得率的影響。在此基礎之上,采用正交試驗法優化提取工藝。此外,本文還采用硅膠柱層析和半制備高效液相分離純化得到高純度的4-HD和XAG,并通過液質聯用儀(HPLC-MS)及核磁共振儀(NMR)對 4-HD及XAG的分子結構及純度進行鑒定。為4-HD、XAG及明日葉的利用奠定了基礎。
1 材料與方法
1.1 材料與儀器
明日葉根:江蘇百代蘭花股份有限公司;硅膠(200~300目):國藥集團化學試劑有限公司;甲醇、乙醇、丙酮、乙酸乙酯、乙醚:均為分析純,國藥集團化學試劑有限公司;液相甲醇:色譜純,百靈威科技有限公司。
Waters1525液 相 色 譜 儀 (PDA檢 測 器Waters2998,自動進樣器 Waters2707):美國 Waters公司;Aduance III 400 MHz核磁共振波譜儀:德國布魯克AXS有限公司;MALDI SYNAPT MS液相色譜串聯質譜聯用儀:美國Waters公司;Waters1525半制備高效液相色譜儀 (PDA檢測器Waters 2998和自動進樣器 Waters 2707):美國 Waters公司;CHRIST冷凍干燥機 ALPHA1-2LD PLUS:德國Marin Christ公司;RV06-ML1B旋轉蒸發儀 (傾斜式):德國 IKA。
1.2 4-HD和XAG含量的測定
1.2.1 混合標準溶液配置 精確稱取4-HD和XAG各5.0 mg,用甲醇溶解并定容于5 mL容量瓶中,得質量濃度為1 mg/mL的混合對照品儲備液。按梯度稀釋成質量濃度分別為 0.6、0.3、0.15、0.12、0.09、0.06及0.03 mg/mL的混合標準溶液。
1.2.2 HPLC色譜條件 色譜柱Phenomenex(C18,4.6 mm×l50 mm,5 μm);柱溫 30 ℃;流動相甲醇∶水=8∶2;流速 1 mL/min,進樣量 20 μL;檢測波長330 nm[13]。
1.3 4-HD和XAG提取條件優化
1.3.1 原料的預處理 新鮮的明日葉根,清水洗滌,除去泥土等雜質,于50℃的電熱恒溫鼓風干燥箱中干燥,粉碎過60目篩,備用。
1.3.2 提取試劑的選擇 準確稱取5.0 g明日葉根粉于圓底燒瓶中,置于水浴鍋中提取,研究提取試劑(乙醚、乙酸乙酯、丙酮、乙醇、甲醇和水)對4-HD和XAG提取得率的影響。
1.3.3 提取單因素試驗 準確稱取5.0 g明日葉根粉于圓底燒瓶中,置于水浴鍋中回流提取,研究乙醇體積分數、溫度、時間、料液比和提取次數對4-HD和XAG提取得率的影響。粗提液旋轉蒸發除去溶劑后,冷凍干燥。將凍干的粗提物用甲醇定容于250 mL的容量瓶中,HPLC分析,以4-HD和XAG的得率為評價指標,確定提取工藝條件。
1.3.4 正交試驗設計 以上討論了各單因素的影響,但實際操作中各因素相互交叉影響。根據單因素試驗結果,選取影響較大的水平進行正交試驗,以確定最佳提取條件。
采用 L9(34)正交表,以提取溫度(A)、乙醇體積分數(B)、料液比(C)、 提取時間(D)作為 4 個考察因素,選取其最佳條件范圍內的3個水平進行試驗。正交設計試驗因素水平見表1。
1.4 4-HD和XAG的純化
稱取明日葉根粉50 g,在最佳條件(料液比1∶10、80%乙醇、提取溫度55℃、提取時間90 min、提取2次)下進行試驗,收集粗提液,減壓濃縮得到深黃綠色浸膏。
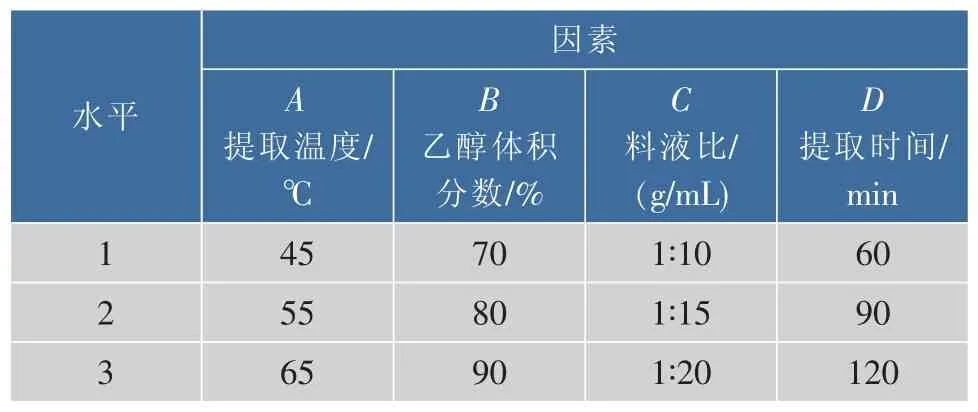
表1 試驗設計因素水平表Table 1 Actors and levels of orthogonal test
往浸膏中加入200 mL去離子水,使其充分溶解,用2倍體積乙酸乙酯萃取4次,合并乙酸乙酯相,減壓濃縮除去乙酸乙酯,得到萃取液浸膏。
浸膏用少量的洗脫劑 (正己烷∶乙酸乙酯=3∶7)溶解,上樣硅膠柱。調節硅膠柱層析的洗脫速度,使其每秒鐘流出1~2滴,每5 min接一管。流出液經HPLC-MS檢測,分別合并含有4-HD和XAG的組分流出液。減壓濃縮除去洗脫劑,得到硅膠柱層析樣品。
用半制備高效液相純化硅膠柱層析樣品。色譜條件如下:半制備 C18柱(10 mm×250 mm,5 μm);柱溫 30 ℃;流動相甲醇∶水=8∶2;流速 4 mL/min,進樣量1 mL;檢測波長330 nm。根據出峰情況分別收集相應組分的流出液。減壓濃縮除去甲醇后冷凍干燥。
1.5 4-HD和XAG的鑒定
對冷凍干燥的樣品進行HPLC-MS及NMR鑒定。其中 HPLC-MS的條件為 CSHC18(2.1mm×100mm,1.7 μm);流動相甲醇∶水=8∶2;柱溫:30 ℃;流速:0.3 mL/min;進樣量:5 μL。質譜條件為離子方式:ESI;毛細管電壓:3.0 kV;錐孔電壓:30 V;離子源溫度:100℃;脫溶劑氣溫度:400℃;脫溶劑氣流速:500 L/h;錐孔氣流速度:50 L/h;碰撞能量(eV):6 V;質量范圍:100~1 500 m/z;檢測器電壓:1 800 V。樣品溶于CDCl3。四甲基硅烷作為化學位移標準品。
2 結果與分析
2.1 4-HD和XAG的混合標準曲線
根據HPLC結果可以得出4-HD和XAG的質量濃度X (μg/mL)與峰面積Y的線性關系擬合標準曲線公式分別為:Y=69 398.19X+136 486.68(R2=0.999 6)、Y=85 102.85X+138 820.79 (R2=0.999 1),線性范圍為0.03~1 mg/mL。混合標準品與樣品的高效液相色譜圖如圖1所示。
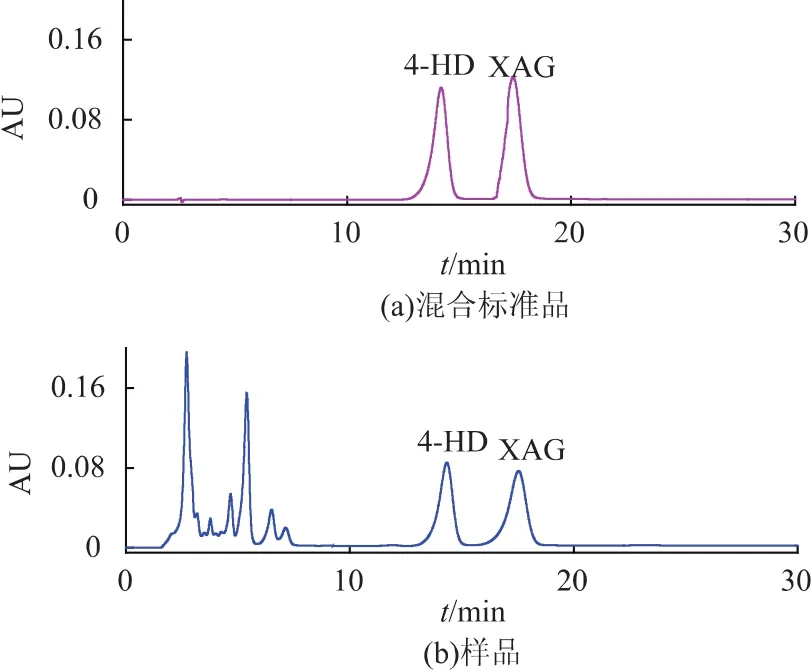
圖1 混合標準品及明日葉根粗提液的HPLC色譜圖Fig.1 HPLC analysis of mixed standard samples and crude extract sample solution
2.2 4-HD和XAG提取條件優化
2.2.1 提取試劑的選擇 試驗結果如圖2所示。從圖中看出,提取試劑對4-HD與XAG得率的影響是一致的。這可能是由于兩種物質具有相似的結構,相似的極性[14]。但不同溶劑對這兩種物質的提取效果差異顯著,乙醇和丙酮的效果較好,甲醇、乙酸乙酯、乙醚次之,水的提取效果最差。這可能是因為乙醇和丙酮的滲透性較大,且與4-HD與XAG的極性較相似,所以乙醇和丙酮的提取效果較好。但是丙酮毒性及揮發性較大,而乙醇較安全,故選擇乙醇作為提取溶劑。
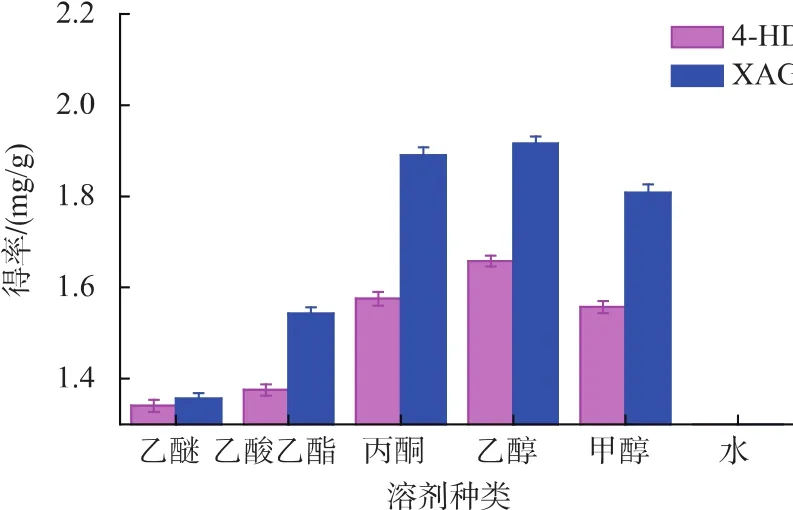
圖2 提取試劑對4-HD和XAG得率的影響Fig.2 Effect of solvent on 4-HD and XAG yield
2.2.2 提取溫度對提取效果的影響 由圖3看出,隨著溫度上升,兩個物質得率逐漸增大,當溫度達到55℃時,這兩物質提取效果最佳。當溫度高于55℃時,這兩物質得率逐漸下降。4-HD和XAG的提取屬于固-液間進行物質傳遞的過程。體系的溫度會影響到原料微粒在溶劑中的運動特性。溫度低時溶劑分子運動慢,不能很好地溶解出4-HD和XAG,溫度升高4-HD和XAG溶出量增加,提取量增大;但對4-HD和XAG的穩定性研究表明,當環境溫度過高時,4-HD和XAG等查爾酮化合物會不可避免地降解[15]。當兩者的作用達到平衡時提取量將不再增加,但當后者的作用占主導地位時提取量就會降低。故提取溫度應選擇55℃左右為宜。
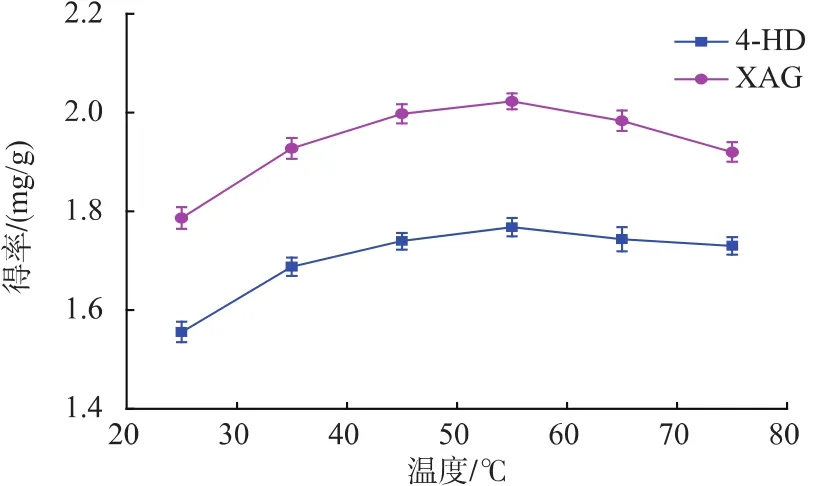
圖3 提取溫度對4-HD和XAG得率的影響Fig.3 Effect of extraction temperature on 4-HD and XAG yield
2.2.3 乙醇體積分數對提取效果的影響 由圖4可知,以乙醇為提取溶劑,體積分數在60%~80%范圍內,得率隨乙醇體積分數的增加而增大,80%乙醇溶液的提取效果最好。80%~100%范圍內,得率隨乙醇體積分數的增加而減小。4-HD和XAG均含有酚羥基,在植物體內會與蛋白質、多糖等以氫鍵及疏水鍵形式結合,而有機試劑具有斷裂氫鍵的作用。但是純的有機試劑不足以破壞氫鍵,故可采用有機試劑和水的復合體系作為提取溶劑。但是4-HD和XAG的極性較低,由“相似相溶”原理知,當提取劑中水的比例過高,使得提取劑極性偏大時,得率會下降。所以乙醇體積分數應選擇80%左右為宜。
2.2.4 料液比對提取效果的影響 料液比增大,傳質動力增加,使原料中更多目標物質進入到溶液中[16]。當浸提劑用量增加到一定程度,傳質達到平衡后,再增加溶劑用量,4-HD和XAG的溶出總量不再增加,而僅僅是更加均勻地分布在溶劑中。考慮到成本,料液比應選擇1∶10左右為宜(圖5)。

圖4 乙醇體積分數對4-HD和XAG提取效果的影響Fig.4 Effect of ethanol concentration on 4-HD and XAG yield
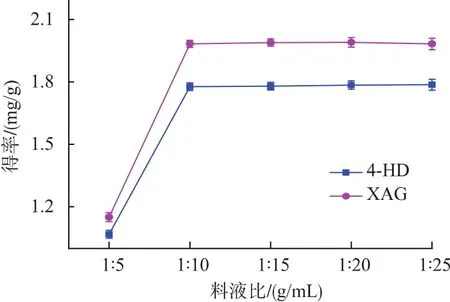
圖5 料液比對4-HD和XAG提取效果的影響Fig.5 Effect of ratio of solid to liquid on 4-HD and XAG yield
2.2.5 提取時間對提取效果的影響 由圖6可知,隨著提取時間的延長,得率先增高,后略微下降。在90 min時,兩物質得率達到最大。提取的傳質過程要有足夠的時間達到平衡,時間太短,提取還沒有達到平衡,提取量就較低;但是如果提取時間過長,4-HD和XAG在較高溫長時間作用下會受到破壞。故提取時間應選擇90 min左右為宜。
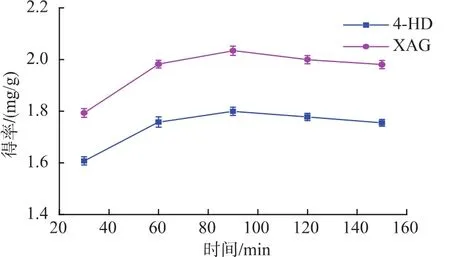
圖6 提取時間對4-HD和XAG提取效果的影響Fig.6 Effect of extraction time on 4-HD and XAG yield
2.2.6 提取次數對提取效果的影響 從圖7可以看出,對4-HD第1次和第2次提取量分別占總提取量的75.21%和22.87%,第3次及第4次提取量分別占總提取量的1.69%和0.23%。對XAG第1次和第2次提取量分別占總提取量的77.80%和19.81%,第3次及第4次提取量分別占總提取量的1.78%和0.61%。總的來說,4-HD和XAG前兩次的提取量分別已達到總提取量的98.08%及97.61%。提取的次數越多,目標產物得率也越多,但同時提取劑消耗量也越多。故提取次數應選擇提取2次為宜。
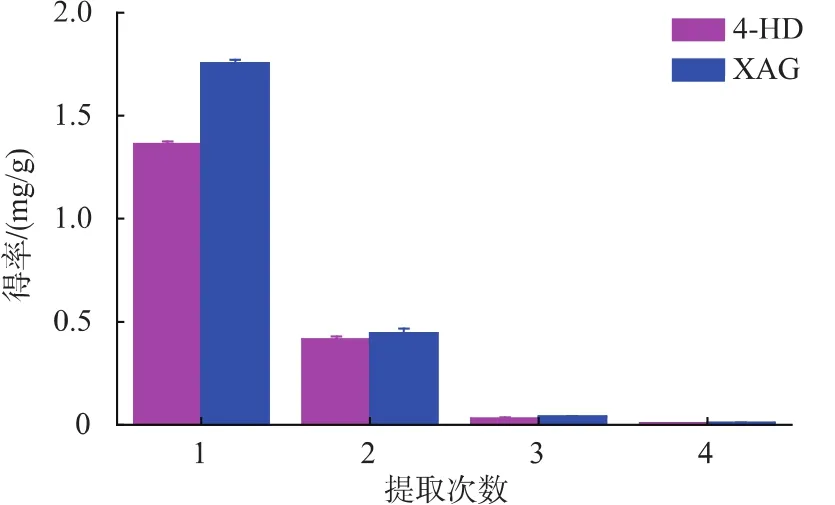
圖7 提取次數對4-HD和XAG提取效果的影響Fig.7 Effect of times of extraction on 4-HD and XAG yield
2.2.7 正交試驗結果與分析 由單因素試驗得,各因素對4-HD與XAG得率的影響是一致的,所以為了方便,在正交試驗中只需考察各因素對4-HD得率的影響情況。正交試驗設計及結果如表2所示。
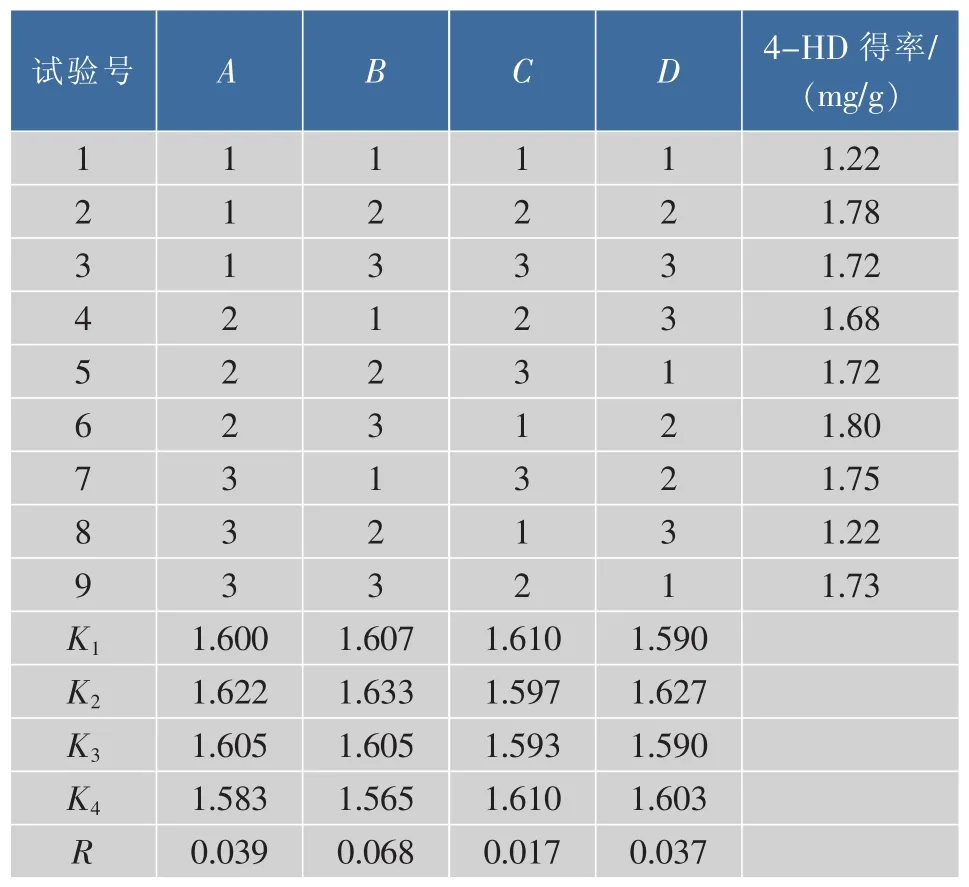
表2 正交試驗設計及結果Table 2 Orthogonal test and results
根據表2中K值可知,最佳提取工藝為A2B2C1D2,即提取溫度55℃,乙醇體積分數80%,料液比1∶10,提取時間90 min。由極差R的大小,可判斷出影響得率的因素順序依次為B>A>D>C,即乙醇體積分數>提取溫度>提取時間>料液比。
2.2.8 驗證試驗 按提取溫度55℃,80%乙醇,料液比 1∶10,提取時間90 min,提取2次進行3次平行試驗,4-HD得率均值為1.82 mg/g,XAG得率均值為2.12 mg/g,高于表2每一項試驗結果,故A2B2C1D2為最佳提取工藝。
2.3 4-HD和XAG的純化
50 g明日葉根粉在最優條件下提取,粗提液冷凍干燥得到13.79 g粗提物,經測定,其中含有91 mg 4-HD、106 mg XAG。考慮到4-HD和XAG極性較低,經乙酸乙酯萃取可進入到乙酸乙酯相中[13],籍此可以初步除去極性較強的雜質,如類黃酮、極性色素、多糖、無機離子等。減壓濃縮除去乙酸乙酯,得到1.03 g萃取物。經萃取后,4-HD的純度由粗提取中的0.66%提高到8.83%;XAG的純度由0.77%提高到10.29%。
萃取物過硅膠柱層析,經洗脫劑(正己烷∶乙酸乙酯=3∶7)洗脫,用HPLC-MS檢測各收集管4-HD和XAG。結果表明,34-41管主要含有4-HD,50-75管主要含有XAG組分(如圖8所示),純度分別為70.63%和67.94%。合并洗脫組分,濃縮除去洗脫劑,得到硅膠柱層析樣品。
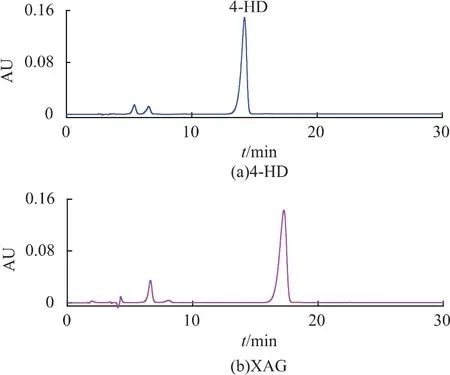
圖8 萃取物經硅膠柱層析洗脫HPLC譜圖Fig.8 HPLC analysis of different fractions purified by silica gel column chromatography
硅膠柱層析樣品用半制備高效液相進行純化。根據出峰情況分別收集相應組分的流出液,分別在22.306和 28.202 min收集到化合物1和化合物2。減壓濃縮除去收集液中的甲醇后冷凍干燥。
2.4 4-HD和XAG的鑒定
對用制備型高效液相純化得到的化合物1和化合物2進行HPLC-MS及NMR結構及純度鑒定。質譜圖如圖9所示。
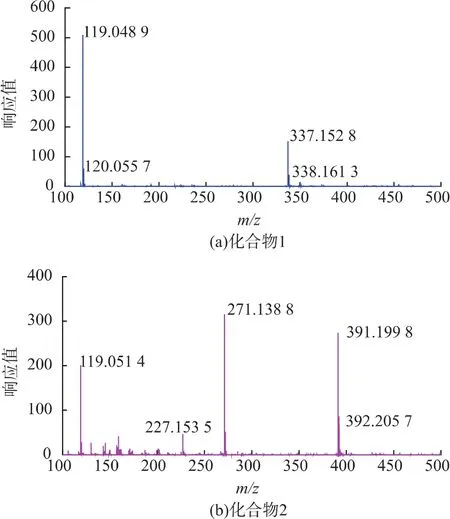
圖9 化合物1和化合物2的ESI-MS圖譜Fig.9 Results of ESI-MS chromatogram of compound 1 and 2
化合物 1質譜數據(圖 9(a)),碎片 m/z 337.152 8是[M-H]-分子離子峰,338.1613是母離子峰,所以其分子量為338。化合物2質譜數據(圖9(b)),碎片m/z 391.199 8是[M-H]-分子離子峰,392.205 7是母離子峰,所以化合物2的分子質量為392。
化合物 1 的 NMR 數據:1H-NMR δ1.71(3H,s,H-5″),1.82 (3H,s,H-4″),3.41 (2H,d,3J(H,H) =7.13 Hz,H-1″),3.93 (3H,s,4′-OCH3),5.26(1H,m,3J(H,H) =7.16 Hz,H-2″),6.52(1H,d,3J(H,H) =9.12 Hz,H-5′) 6.90(2H,d,3J(H,H) =8.44 Hz,H-3,5),7.48(1H,d,3J(H,H) =15.45 Hz,H-α),7.56(2H,d,3J (H,H) =8.35 Hz,H-2,6),7.81(1H,d,3J(H,H) =9.29 Hz,H-6′),7.84(1H,d,3J(H,H)=16.60 Hz,H-β),13.48 (1H,s,2′-OH)。13C-NMR δ 17.84 (C-4″),21.75 (C-1″) 25.81 (C-5″),55.80(OCH3),102.21(C-5′),114.67(C-1′),116.08(C-3,5),117.62(C-3′),118.08(C-α),122.05(C-4′),127.63(C-1),129.25(C-5′),130.58(C-3″),131.97(C-2,6),144.19 (C-β),158.24 (C-4),162.99 (C-2′),163.35(C-4′),192.53(C=O)。
化合物 2 的 NMR 數據:1H-NMR δ1.59(3H,s,H-9″),1.67(3H,s,H-8″),1.83(3H,s,H-10″),2.10(4H,m,H-4″,5″),3.49(2H,d,3J(H,H) =7.14 Hz,H-1″),5.06(1H,t,3J(H,H)=6.09 Hz,H-6″),5.30(1H,dd,3J (H,H) =7.20 Hz,H-2″),6.43(1H,d,3J(H,H) =8.90 Hz,H-5′),6.88 (2H,d,3J (H,H) =8.46 Hz,H-3,5),7.46(1H,d,3J(H,H) =15.44 Hz,H-α),7.57(1H,d,3J(H,H)=8.58 Hz,H-2,6),7.57(1H,d,3J (H,H) =8.90 Hz,H-6′),7.86(1H,d,3J(H,H)=15.37 Hz,H-β),13.88(1H,s,2′-OH).13CNMR δ 16.31(C-10″),17.70(C-9″),21.81(C-1″),25.63(C-8″),26.48(C-5″),39.77(C-4″),114.20(C-1′,3′),116.11(C-3,5),118.38(C-α),121.11(C-2″),123.83 (C-6″),127.99 (C-1),129.29 (C-6′),130.56 (C-2,6),132.07 (C-7″),139.73 (C-3″),144.09(C-β),158.08(C-4),161.86(C-4′),163.95(C-2′),192.36(C=O)。
所以由此確定化合物1和化合物2的分子分別為C21H22O4、C25H28O4。結構式見圖10。即化合物1為4-HD,化合物2為XAG。
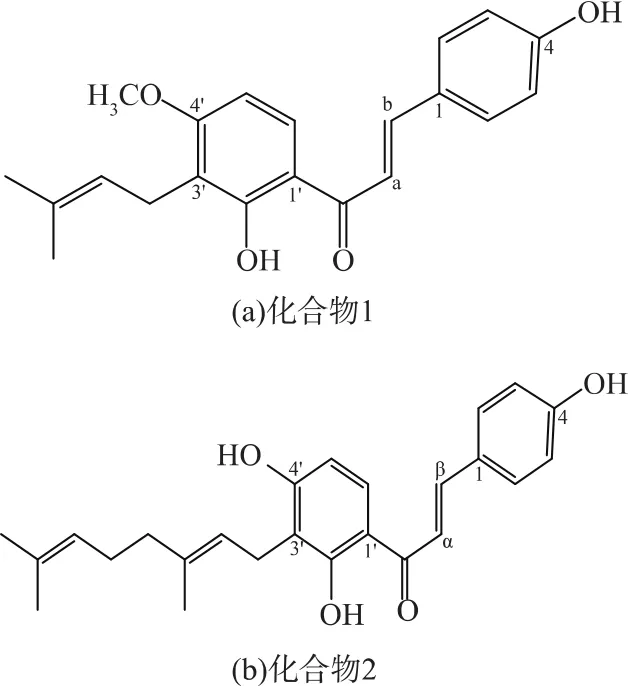
圖10 化合物1和化合物2的結構式Fig.10 Chemical structures of compound 1 and 2
3 結 語
本文以明日葉為原料,首次對4-羥基德里辛和黃色當歸醇的提取工藝進行探討。通過單因素以及正交試驗得出4-羥基德里辛和黃色當歸醇最佳提取工藝,條件為:提取次數2次、提取溶劑80%乙醇、提取溫度55℃、提取時間90 min、料液比 1∶10。在此條件下,4-HD得率為1.82 mg/g,XAG得率為2.12 mg/g。這為明日葉以及4-羥基德里辛和黃色當歸醇的利用奠定了基礎。采用乙酸乙酯萃取、硅膠柱層析以及半制備液相相結合的方法對粗提液進行純化,經HPLC-MS及NMR分析表明,純化得到的兩種物質為4-羥基德里辛和黃色當歸醇,純度分別為99.08%和98.92%。
