吳茱萸次堿:一種多靶點天然化合物的藥理學作用研究進展
2019-01-03 01:30:46羅丹,顏行
天然產物研究與開發 2018年12期
羅 丹,顏 行
1南昌大學醫學院生理學系;2南昌大學藥學院基礎藥理重點實驗室,南昌330006
大量臨床研究發現,作用于單靶點的藥物在治療動脈粥樣硬化、糖尿病、腫瘤等發病機制復雜的疾病時常療效不佳。而多靶點藥物可同時作用于疾病發病機制中的多個靶點,產生協同效應,減少副作用和抗藥性的產生,從而達到更佳療效。中藥有效成分往往通過多個靶點而發揮系統治療作用,是我們設計先導化合物尋找多靶點藥物的寶貴資源。傳統中藥吳茱萸屬蕓香科植物,始載于《神農本草經》,臨床應用已有數千年歷史。吳茱萸次堿(rutaecarpine,Rut)是從吳茱萸將近成熟的果實中提取出來的一種吲哚喹啉類生物堿,其化學結構式為C18-H13-N3-O。作為吳茱萸的主要活性成分之一,Rut具有廣泛的藥理作用[1],如心血管保護作用、改善腦功能、保護胃黏膜、抗炎抗氧化等,其藥理學作用機制涉及多種生物靶點。本文對近十年來文獻報道的Rut的作用的靶點及藥理學作用,及以Rut為先導化合物開發的衍生物及類似物的相關進展進行綜述,為Rut對相關疾病的治療提供依據。
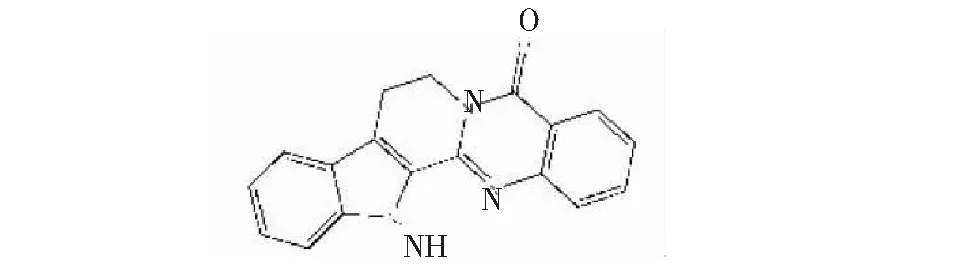
圖1 吳茱萸次堿的化學結構Fig.1 The chemical structure of rutaecarpine
1 Rut的作用靶點
1.1 TRPV1
辣椒素受體(Transient receptor potential vanilloid 1,TRPV1),屬瞬時感受器電位離子通道(TRP)超家族中的成員,是一種多型信號探測器,可被高溫、氫離子等理化因素及天然配體如辣椒素、Rut、樹脂毒素等激活。TRPV1為一種配體門控的非選擇性陽離子通道,激活可產生以Ca2+內流為主的陽離子內流。TPPV1主要分布于感覺神經元末梢,也廣泛存在于非神經組織,如血管內皮細胞、平滑肌細胞、單核細胞等。國內外大量研究提示,Rut為TRPV1的激動劑,其多種藥理學效應均通過TRPV1介導。最早日本學者Kobayashi在豚鼠的心房肌發現, Rut對心肌有正性肌力和正性頻率作用,該效應可被TRPV1非選擇性的阻斷劑釕紅所取消;隨后Hu和Deng等的研究相繼證實Rut的心臟保護作用及舒張血管和降壓作用均與激活TRPV1有關,其效應可被TRPV1競爭性阻斷劑Capsazepine所阻斷[1]。最近Wang等的研究提供了直接證據,Rut及吳茱萸的另一有效成分吳茱萸堿與辣椒素有相同的結合位點,是TRPV1的部分激動劑[2]。他們采用膜片鉗的技術在轉染TRPV1受體的HEK293細胞證實,Rut及Evo能激活TRPV1介導的陽離子內流,兩者的效應分別比辣椒素低9倍和3.5倍。值得注意的是,辣椒素和吳茱萸堿等TRPV1激動劑反復使用均會產生受體脫敏,而Rut不會產生這種脫敏作用。這提示Rut作為TRPV1部分激動劑,通過適度激活TRPV1產生效應,可能具有更為廣泛的臨床應用前景。
1.2 NADPH氧化酶
煙酰胺腺嘌呤二核苷酸磷酸(Nicotinamide-adenine dinucleotide phosphate,NADPH)氧化酶是體內活性氧 (Reactive oxygen species,ROS)產生的主要來源之一。Heo等[3]在培養的單核細胞發現, Rut可抑制致動脈粥樣硬化因子LIGHT誘導的NADPH的激活和ROS的產生,進而抑制炎癥因子的釋放和單核細胞遷移。隨后,Bao等[4]在心肌細胞的缺氧復氧模型也證實,Rut可抑制NADPH氧化酶的活性,下調NADPH氧化酶亞基Nox2,Nox4,及調節蛋白 p47(phox)表達上調而減少ROS的生成。這些研究提示,Rut可能通過抑制NADPH氧化酶源性的ROS生成而發揮抗氧化應激作用。
1.3 環氧化酶-2
環氧化酶(Cyclooxygenase,COX)是催化花生四烯酸轉化為前列腺素(prostaglandins,PGs)的關鍵酶,主要包括COX-1和COX-2兩種同工酶。前者為結構型,主要存在于正常的組織細胞中,催化產生維持正常生理功能的PGs。后者為誘導型,可被各種病理因素誘導表達,產生大量PGE2,介導炎癥反應。因而,COX-2是目前非甾體抗炎藥的主要作用靶點。Moon等的研究提示,Rut可能為一種新型的COX-2抑制劑。在骨髓源性的肥大細胞,Rut劑量依賴性地抑制COX依賴性的PGD2生成,在傳染了COX-2的HEK293細胞,Rut可抑制外源性花生四烯酸轉化為PGE2。Rut并不能抑制COX-1的活性和COX-2的蛋白和mRNA表達水平,提示其可能通過直接抑制COX-2的活性發揮抗炎效應。最近Lee等在培養的小鼠巨噬細胞株RAW264.7中發現,Rut的低毒衍生物Br-Rut可顯著抑制脂多糖誘導的COX-2蛋白水平升高[5]。
1.4 膽堿酯酶
乙酰膽堿酯酶(acetylcholinesterase,AChE)是體內誘導乙酰膽堿水解失活的關鍵酶。He等[6]的研究報道,Rut的衍生物表現為高選擇性的AChE抑制作用。通過抑制雙倒數作圖法和分子模型研究結果表明,這些化合物可以分別和AChE的催化活性區及外周陰離子區結合,抑制AChE的水解而增加內源性乙酰膽堿的水平。
1.5 細胞色素P450
細胞色素P450(cytochrome P450,CYP450)是調節內源性和外源性物質(如藥物、環境化合物等)代謝的關鍵酶。大量研究表明Rut可影響CYP450的作用,從而影響藥物的代謝過程。這一效應在中藥及藥物的相互作用中值得注意。在大鼠和小鼠肝臟微粒體的研究發現Rut可誘導 CYP450 1A 和2B。Rut可提高CYP1A2的催化活性,從而促進氨茶堿的分解代謝,兩藥合用可降低氨茶堿的藥效[7]。但也有報道認為Rut可抑制某些CYP酶的活性,如在小鼠和人肝微粒體的研究發現,吳茱萸次堿是一種CYP1A2的選擇性抑制劑。最近研究認為Rut的肝臟毒性及誘導藥物相互作用可能與抑制CYP1A2,CYP3A4等多種CYPs的活性有關[8]。
1.6 β1腎上腺素能受體
Xue 等[9]結合β1腎上腺能受體/細胞膜層析技術及超高液相色譜/質譜技術篩選發現,Rut是β1受體的拮抗劑,離體實驗證實Rut下調β1受體下游cAMP和PKA的水平,在體實驗發現Rut通過抑制β1受體調節能量代謝可抑制心肌缺血再灌注損傷。
1.7 膽固醇逆向轉運蛋白ABCA1
膽固醇逆向轉運蛋白(ATP-binding cassette transporter A1,ABCA1)是促進膽固醇外排的關鍵轉運體。Li等[10]的系列研究發現Rut及其衍生物可上調ABCA1的表達,提示Rut具有抗動脈粥樣硬化的潛在藥理學作用。
2 Rut的藥理學作用
2.1 心臟保護作用
在多種心臟缺血模型中均證實Rut具有顯著的心臟保護效應,其主要機制均與激活TRPV1受體促進降鈣素基因相關肽(Calcitonin gene-related peptide,CGRP)釋放有關。CGRP是辣椒素敏感的感覺神經的主要遞質,廣泛分布于神經和心血管系統中,是一種重要的內源性心肌保護物質。離體心臟灌流和在體實驗均發現,Rut可減輕缺血再灌注所致心肌損傷,增加冠脈流出液或血清中CGRP的水平,其心臟保護效應可被競爭性TRPV1受體阻斷劑Capsazepine和選擇性CGRP受體阻斷劑CGRP8-37 取消[1]。此外,Rut還可有效逆轉多種病理因素誘導的心臟肥厚。異丙腎上腺素誘導的心臟重構模型中發現,Rut可逆轉心臟重構,表現為降低左心室與體重比及心臟橫截面積,抑制心肌細胞凋亡和膠原沉積,其機制與上調CGRP水平有關[11]。最近Li等[12]發現,Rut可抑制低氧誘導的右室重構,減少TGF-b誘導的心肌成纖維細胞的增殖,其機制與促進CGRP釋放,進而調節eIF3a/p27通路有關。
2.2 對血管的影響
2.2.1 舒張血管和降壓
中藥吳茱萸內服和外敷均能有效降低血壓,其主要成分Rut在離體血管和整體動物水平均證明能有效地舒張血管和降壓,可能涉及以下機制: Wang等[13]研究認為,Rut可促進內皮細胞非電壓依賴性鈣通道開放,升高胞內鈣水平,后者通過NO-cGMP途徑而發揮內皮依賴性舒張血管作用;而在培養的血管平滑肌細胞,Rut則抑制L型電壓依賴性Ca2+通道開放,從而導致Ca2+內流減少,血管平滑肌舒張。Li課題組[14]系列研究則表明Rut的舒血管和降壓效應是通過激活TRPV1促CGRP合成和釋放所介導的,后者是一種強大的舒血管活性物質,其舒血管效應比乙酰膽堿強1000倍。主要證據如下:在離體的大鼠血管環和在體實驗中發現,Rut的舒血管和降壓效應均伴隨CGRP濃度的升高。預先給予辣椒素耗竭CGRP和CGRP受體阻斷劑CGRP8-37,可顯著抑制Rut的上述效應。在兩腎一夾型高血壓大鼠[15]和自發性高血壓大鼠模型[16]中均發現,連續口服Rut產生穩定的降壓效應,伴隨血漿CGRP濃度及背根神經節CGRP的mRNA表達水平升高,這些作用可被預先給予辣椒素所減弱。該課題組最近的研究在穩定轉染TRPV1的293細胞和培養的背根神經節細胞發現,Rut可劑量依賴性上調CGRP的表達,該效應可被TRPV1阻斷劑CAPZ所取消,Rut可升高細胞內Ca2+的水平[17]。
2.2.2 保護血管內皮
我們前期的研究發現,吳茱萸次堿可抑制LPC誘導的血管內皮損傷和功能紊亂,其機制與激活TRPV1上調內源性血管保護物質CGRP水平有關[18]。循環中的內皮祖細胞在維持內皮完整和促進血管修復有重要生理學意義。內皮祖細胞的衰老和功能失調與多種心血管疾病有密切關系。Zhou等[19]研究發現,高血壓狀態下內皮祖細胞的衰老加速與體內CGRP生成減少有關,給予Rut激活TRPV1促進內源性CGRP生成可有效抑制高血壓和AngII誘導的內皮祖細胞衰老,其效應可被CGRP8-37和capsazepine所阻斷。
2.2.3 抑制血管平滑肌增殖
血管平滑肌細胞異常增殖是多種因素誘導血管重構狹窄的重要病理基礎。在培養的大鼠胸主動脈平滑肌細胞發現,Rut可下調VSMC增殖相關基因c-myc的表達,上調eNOS促進NO生成,從而抑制Ang II誘導的血管平滑肌細胞增殖[20]。最近有研究報道[21],Rut可抑制缺氧誘導的人肺動脈血管平滑肌HPASMCs增殖,并誘導其凋亡,其機制與抑制HIF-1α表達及下游信號通路有關,表現為減少PCNA表達,上調p53和 p21的表達。
2.2.4 抗炎
炎癥是機體對內外刺激的一種防御反應,也是多種疾病發生發展的病理生理基礎。Rut在多種炎癥模型中均能起到抗炎作用,可能涉及多種機制。
2.2.3.1 抑制前列素合成
前列腺素PGE2、PGD2在炎癥反應中起著重要作用。在培養的小鼠巨噬細胞株RAW264.7中發現,Rut可顯著抑制脂多糖誘導的PGE2生成,至少部分介導其抗炎機制[22]。如前所述,Rut可通過抑制PGs合成的限速酶COX- 2而減少前列腺素生成。然而,Woo等在鈣離子載體A23187誘導的小鼠巨噬細胞炎癥模型發現,Rut可直接抑制合成PGs的前體花生四烯酸釋釋放,但對COX-1和COX-2活性卻無顯著影響[23]。
2.2.3.2 抗氧化
眾所周知,ROS過度生成誘導基因表達的改變在炎癥反應中亦起著重要作用。NADPH氧化酶是體內生成ROS的重要限速酶。日本學者等發現Rut可抑制LIGHT誘導的單核細胞遷移,其機制與抑制NADPH的激活導致的ROS的產生,進而抑制CCR1、CCR2、ICAM-1、 ERK和p38 MAPK等炎癥過程[3]。最近Jin等[24]研究發現,Rut可抑制叔丁基過氧化氫誘導的肝細胞損傷,其機制涉及激活CaMKII-PI3K/Akt-Nrf2抗氧化信號途徑,上調其下游靶基因血紅素氧合酶-1hemeoxygenase-1 (HO-1)HO-1表達,進而抑制AST,ALT和脂質過氧化有關。
2.2.4 抗動脈粥樣硬化
大量研究表明Rut具有保護血管內皮、抑制單核細胞遷移粘附、調節血脂、抑制血栓形成等多種抗AS效應,可能是一種潛在的抗AS藥物。
2.2.4.1 抑制內皮-單核細胞粘附
循環中的單核細胞與受損的血管內皮粘附,遷移并吞噬脂質發展為泡沫細胞,是動脈粥樣硬化斑塊形成的關鍵環節。Rut可抑制致AS因子LIGHT誘導的單核細胞遷移及IL-8、MCP-1、TNF-α等多種炎癥介質的釋放[3]。近年來大量研究提示縫隙連接蛋白(connexin,Cx)與動脈粥樣硬化密切相關,參與介導單核細胞和血管內皮的相互作用。其中Cx37和Cx40被認為有抗AS作用,而Cx43過度表達則有促AS效應。我們最近的研究表明[25,26],Rut可抑制Ox-LDL誘導的內皮功能紊亂和單核細胞粘附,促進內皮源性Cx37和Cx40及單核源性Cx37的表達,抑制兩者Cx43的表達,其機制與激活TRPV1有關。
2.2.4.2 促進膽固醇排出
Yu等[27]研究提示,Rut可通過促進膽固醇外排而抑制AS斑塊形成。離體水平, Rut可促進巨噬細胞株RAW264.7中膽固醇外流。在ApoE-/-小鼠,給予Rut八周可顯著抑制斑塊面積,減少斑塊區的膽固醇和巨噬細胞聚集,其機制與促進肝臟中膽固醇逆向轉運蛋白ABCA1 和清道夫受體SR-BI/CLA-1的表達有關。
2.2.4.3 抗血栓作用。
血栓形成是動脈粥樣硬化斑塊形成的終末病理變化。離體和在體的實驗均證明Rut可抑制多種因素(如膠原、ADP、腎上腺素和花生四烯酸)誘導的血小板聚集和血栓形成[28]。在小鼠腸系膜上靜脈血栓模型,Rut可以顯著抑制血小板血栓形成,表現為延長靜脈的阻塞時間、出血時間和血小板血栓的潛伏期,其作用甚至超過陽性對照藥阿司匹林。此外,靜脈注射Rut亦能有效降低ADP誘導的小鼠急性肺血栓栓塞癥的死亡率。其抗血小板活化機制可能通過抑制了磷脂酶C的活性,導致磷酸肌醇的分解減少,使血栓烷素A2的生成減少,進而抑制血小板內Ca2+動員[29]。此外,Li等[30]研究認為,Rut抑制血小板聚集的作用與上調CGRP表達有關,后者可抑制血小板源性組織因子TF的釋放。
2.3 抑制肥胖與代謝綜合癥
在高脂飲食誘導和瘦素缺陷兩種肥胖小鼠模型的研究發現,Rut可通過抑制攝食而降低肥胖,并降低血脂、血糖和胰島素水平,其機制與抑制下丘腦中兩種具有促進食欲作用的神經肽(神經肽Y和刺鼠色蛋白相關蛋白)的表達有關[31]。Chen等[32]研究發現,Rut及其衍生物可抑制脂肪細胞的分化和脂質合成,減少脂質堆積,其機制與激活AMPK信號通路,抑制非折疊蛋白UPR通路有關。最近在高脂飲食誘導的STZ糖尿大鼠的高脂高糖模型發現,Rut可顯著抑制肥胖和內臟脂肪素堆積,減低血脂和血糖,增高胰島素敏感性。降低肝臟中的NF-KB活性,抑制IL-6,CRP and MCP-1等炎癥因子的釋放,減輕肝臟和胰臟的病理損傷。前者與通過肝臟的IRS-1/PI3K/Akt通路有關,后者通過AMPK/ACC2促進糖攝取[33]。這些研究提示,Rut具有潛在的減肥和抗糖尿病代謝綜合征效應。
2.4 抗阿爾茨海默病
阿爾茨海默病(Alzeheimer’s,AD)俗稱老年性癡呆,是一種進行性發展的神經系統退行性疾病。乙酰膽堿為促進學習記憶的重要神經遞質,老年癡呆的重要病理機制之一就是膽堿能神經元的退化。膽堿酯酶抑制劑(AChE)可通過抑制乙酰膽堿的降解而提高內源性乙酰膽堿的水平,因而成為老年癡呆治療最常用的一類藥物。最近有研究報道[6],Rut的低毒衍生物7,8-脫氫Rut是膽堿酯酶的選擇性抑制劑,能減少β-淀粉樣蛋白聚集,再加上強大的抗氧化和金屬離子螯合作用,提示其可被開發成新型的多功能抗AD藥物。
2.5 胃粘膜保護作用
中藥吳茱萸治療胃腸道紊亂已有數千年歷史。胃腸道分布有豐富的辣椒素敏感的感覺神經,其主要遞質CGRP是一種重要的內源性胃黏膜保護物質,機制涉及促進胃黏膜血流,抑制胃酸分泌,減少胃黏膜細胞的凋亡和氧化損傷等[34]。在阿司匹林[35]和酒精[36]誘導的大鼠胃潰瘍模型的研究均發現,Rut可顯著降低潰瘍指數和H+返流,其機制與激活TRPV1促進CGRP釋放有關。
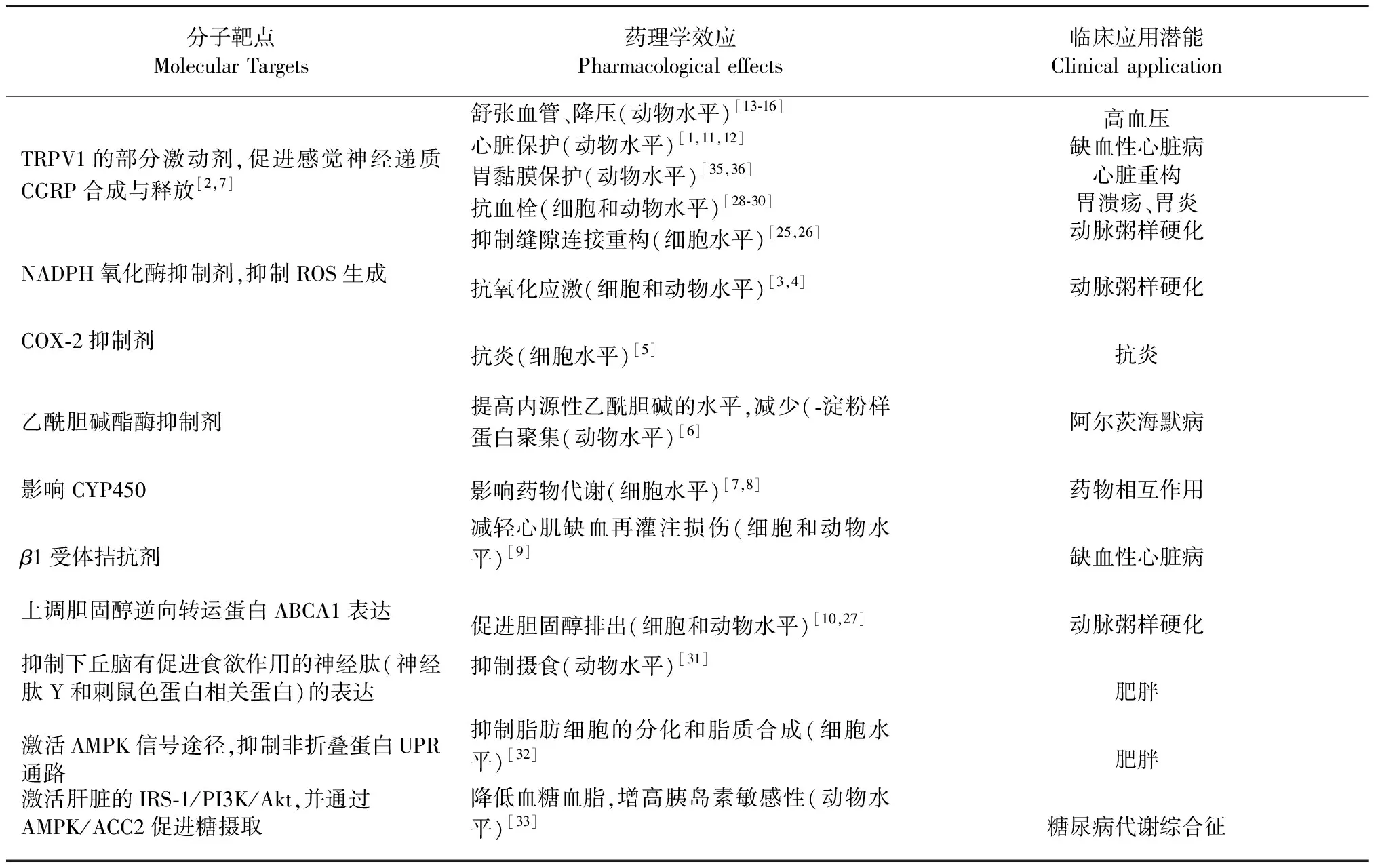
表1 吳茱萸次堿的主要作用靶點和臨床應用Table 1 The major targets and clinical applications of rutaecarpine
3 吳茱萸次堿的衍生物和類似物
目前吳茱萸次堿主要通過化學合成或生物提取的方法獲得。大量研究通過對吳茱萸次堿結構進行化學修飾,開發出一系列吳茱萸次堿的衍生物和類似物,可改變其藥理學活性或理化特性,這些構效關系的研究有助于開發新的選擇性更高藥效更穩定的天然藥物[37]。
在研究Rut及其衍生物的抗血小板活性作用發現,2,3-亞甲二氧基吳茱萸次堿,3-氯代吳茱萸次堿和3-羥基吳茱萸次堿在低濃度就表現為更強的抑制血小板聚集的作用。3-甲氧基吳茱萸次堿的丁酸衍生物還可抑制ADP、膠原等誘導的血小板聚集。這些衍生物均表現為優于吳茱萸次堿的抗血小板活性[38]。
Chen等[39]研究比較了12種Rut的衍生物和11種類似物的舒血管活性,發現N14原子可能是關鍵部位,該位點的修飾可增加舒血管活性,而5-羰基的作用不大,單純吲哚或喹唑啉環的取代也不能增加舒血管活性。
在Rut衍生物的抗腫瘤作用的研究發現,不同位點的環取代可導致對不同的腫瘤細胞的選擇性毒性。如11-甲氧基吳茱萸次堿表現為對肺癌和腎癌細胞的細胞毒性。10,11-亞甲二氧基 類似物則表現為對卵巢癌細胞的細胞毒性[40]。Rut衍生物的抗腫瘤細胞毒性機制與抑制拓撲異構酶有關,而對拓撲異構酶I和II的抑制作用可能與E環取代有關,如10-溴代吳茱萸次堿和 3-氯代吳茱萸次堿均表現為較強的對拓撲異構酶I和II的抑制作用[41]。
Wang等[42]研究了一系列Rut衍生物對膽堿酯酶的抑制性。這些衍生物的抗膽堿酯酶活性及對乙酰膽堿酯酶(AChE)或丁酰膽堿酯酶(BuChE))的選擇性抑制作用可能與骨架結構及側鏈的長度有關:如基本骨架中由芳香C環的衍生物較無芳香環及開鏈結構的衍生物有更強的活性和選擇性;7,8-去氫吳茱萸次堿的衍生物較吳茱萸次堿的衍生物及開C環衍生物表現為更強的AChE的高選擇性及抑制效應;此外,提高側鏈的長度也增加衍生物的選擇性和活性[6]。
4 展望
對天然藥物有效成分的多靶點調節作用的研究將成為探索中藥作用機制,挖掘中藥新的臨床應用的重要趨勢。近年來,吳茱萸的另一個有效成分吳茱萸堿的多靶點抗腫瘤效應倍受關注,其可影響多條信號轉導通路而抑制腫瘤細胞的增殖、侵襲、轉移和血管新生,有望開發為新的多靶點抗腫瘤藥物[43]。Rut的抗腫瘤效應較吳茱萸堿弱,但在心腦血管領域有較好臨床應用前景。目前研究認為Rut的較多藥理學作用均與激活TRPV1有關,后者通過促進鈣內流可觸發一系列生理學效應。Rut對其他靶點的作用是TRPV1激活胞內鈣信號途徑的下游機制,還是獨立于TRPV1之外的直接作用,尚需研究。并且目前作用靶點大多采用藥理學方法推測,其蛋白相互作用及構效關系尚未完全闡明。此外,RUT的低溶解度也是影響其作為藥物的生物利用度的重要原因,因而Rut在離體實驗水平效果較好,而在整體水平灌胃給藥效果欠佳。因而,以吳茱萸次堿為分子骨架,結合計算機分子模擬、基因敲除和藥物化學等技術對吳茱萸次堿進行結構創新和靶點確證,明確其構效關系,升高生物活性和生物利用度,降低毒性,是開發吳茱萸次堿這一多靶點藥物的趨勢。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
四川勞動保障(2021年9期)2022-01-18 05:11:08
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
文苑(2018年21期)2018-11-09 01:23:06
汽車工程學報(2017年2期)2017-07-05 08:13:02
中國衛生(2016年9期)2016-11-12 13:28:08
中國衛生(2015年9期)2015-11-10 03:11:12
